
Болезнь Фабри
| Классификация по МКБ-10 | |
|---|---|
| E75.2 | Другие сфинголипидозы (включая болезнь Фабри (Андерсона)) |
| МКБ-10 онлайн (версия ВОЗ 2019 г.) | |

Мутация в гене GLA , который кодирует этот белок, вызывает снижение активности этого фермента у пациентов с болезнью Фабри. В результате некоторые жиры (гликосфинголипиды) не могут быть расщеплены в достаточной степени и накапливаются в разных клетках. Эти отложения приводят к симптомам болезни Фабри.
Болезнь Фабри (также называемая болезнью Фабри , синдром Фабри или болезнью Фабри-Андерсон ) является редким, врожденным , моногенным нарушением обмена веществ , принадлежащим к группе лизосомных болезней накопления . У пораженных пациентов отсутствует фермент (катализатор). Болезнь Фабри является наследственной и поддается лечению.
Болезнь Фабри - это мультисистемное заболевание, которое может поражать большое количество органов в организме. Симптомы могут сильно различаться в зависимости от пораженных органов . Индивидуально очень разные проявления болезни и ее редкость значительно усложняют диагностику ; обычно это делается правильно только через много лет после появления первых симптомов. Заболевание в основном поражает мужской пол, но могут заболеть и гетерозиготные (метисы) женщины. Однако у них болезнь обычно менее выражена и начинает приобретать клиническую значимость только в среднем возрасте. Качество жизни пациентов с болезнью Фабри часто значительно ухудшается.
Заболевание было причинным излечимо с заместительной терапией фермента с 2001 года . Пациенты получают генно-инженерную α-галактозидазу А в виде инфузии на всю оставшуюся жизнь . Болезнь Фабри в настоящее время неизлечима. Если не лечить, пациенты мужского пола достигают среднего возраста около 50 лет, а пациенты - около 70 лет. Основными причинами ранней смертности являются хроническая почечная недостаточность , поражение сердца и нарушение кровоснабжения головного мозга .
Из-за мутации (генетического изменения) в Х-хромосоме (половой хромосоме) активность фермента α-галактозидазы А значительно снижается. Он отвечает за расщепление сахаристых жирных веществ. В лизосомах (центры рециркуляции клеток) продукт метаболизма глоботриаозилцерамид (также называемый Gb3 или GL-3), гликосфинголипид , больше не может быть расщеплен в достаточной степени. Gb3 основном накапливается в клетках на внутренней оболочки в кровеносных сосудах , в эндотелиальных клетках . По мере прогрессирования заболевания эти скопления становятся патологическими , то есть вызывают болезнь Фабри. В зависимости от течения болезни на это могут уйти десятилетия.
Заболевание впервые было независимо описано в 1898 году немцем Иоганном Фабри и англичанином Уильямом Андерсоном .
Эпидемиология
Заболевание поражает все расы , и оба пола могут заболеть этим заболеванием. Их редкость затрудняет точное определение их частоты. В литературе приводятся значения распространенности от 1: 476 000, от 1: 117 000 до 1: 40 000. Однако более поздние исследования, основанные на данных скрининга новорожденных , указывают на гораздо более высокую заболеваемость болезнью Фабри. В северной Италии 3100 и в: распространенность около 1 2004-2006 гг. Тайвань 1 из около 1: 1500, определенная у новорожденных мужского пола.
Если взять за основу распространенность 1: 3100, это даст 26 450 пациентов с болезнью Фабри в Германии. Официально известно, что в Германии пострадали около 700 человек, а также гораздо большее количество незарегистрированных случаев. Предполагается, что у многих пациентов заболевание не распознается в течение их жизни и что преждевременная смерть связана с другими заболеваниями, например изолированными кардиомиопатиями , как это может происходить у пациентов с болезнью Фабри с остаточной активностью α-галактозидазы А. Другим признаком этого является то, что 86% новорожденных, получивших положительный результат на Тайване, имеют криптическую сплайс-мутацию типа IVS4 + 919G> A, которая ранее была обнаружена в основном у пациентов с болезнью Фабри с сердечным фенотипом. У этих пациентов болезнь Фабри в основном проявляется заболеванием сердечной мышцы (кардиомиопатией). Интронная форма этой мутации встречаются во многих тайваньских больных с гипертрофической кардиомиопатией .
В некоторых субпопуляций , связанных с симптомами болезни Фабри, то заболеваемость , естественно , выше. В исследовании с участием 911 испанских пациентов, находящихся на гемодиализе , у четырех мужчин и трех женщин были выявлены изменения в гене GLA , что соответствует распространенности 1: 182 в этой субпопуляции.
Генетика и молекулярная биология



Болезнь Фабри - это заболевание, в основе которого лежит генетический дефект (мутация) женской половой хромосомы , Х-хромосомы. Любой отец, пораженный этой болезнью, унаследует болезнь всем своим дочерям, в то время как все его сыновья останутся здоровыми. Если мать несет мутировавший ген, ее дети - независимо от пола - имеют 50% риск унаследовать болезнь. Мутация затрагивает ген GLA , который расположен на длинном плече Х-хромосомы в локусе гена q22.1.
Генный продукт представляет собой гомодимер , и, как и все лизосомальных ферментов, является котрансляционно при условии , с манноза-6-фосфата остатка при переводе мРНК в аминокислотной последовательности . Часть молекулы фосфорилированной α-галактозидазы A происходит из секретируемых клеток и других клеток через мембраносвязанный маннозо-6-фосфатный рецептор путем эндоцитоза . Возобновление фосфорилированной α-галактозидазы A через маннозо-6-фосфатный рецептор является основой для заместительной ферментной терапии.
Особенности Х-сцепленного наследования
Из-за наследования, сцепленного с Х-хромосомой, болезнь протекает по-разному у мужчин и женщин. Пациентов мужского пола называют гемизиготными, а женщин - гетерозиготными носителями. Раньше считалось, что только у мужчин может развиться болезнь Фабри, а гетерозиготные женщины являются только носителями. Так обстоит дело с подавляющим большинством других наследственных заболеваний, связанных с Х-хромосомой, таких как гемофилия или мышечная дистрофия типа Дюшенна . Теперь известно, что у женщин с такой характерной гетерозиготностью также может развиться болезнь Фабри. Поэтому некоторые авторы рекомендуют избегать термина Х-сцепленный рецессивный , поскольку он вводит в заблуждение. Вместо этого рекомендуется использовать терминологию Х-сцепленное наследование (англ. X-connected наследование ).
У гетерозиготных пациентов имеется одна немутантная и одна мутированная Х-хромосома в каждой ядросодержащей клетке тела с ДНК . За счет X-инактивации одна из двух X-хромосом инактивируется в каждой клетке. Эта инактивация происходит в каждой клетке независимо и по принципу случайности (так называемая мозаика ). С чисто статистической точки зрения, 50% клеток будут вырабатывать α-галактозидазу A с отсутствующей или пониженной активностью - в зависимости от типа мутации. Остальные 50% клеток продуцируют α-галактозидазу А с нормальной активностью («здоровые клетки»). Часть активной α-галактозидазы A подвергается эндоцитозу «мутировавшими клетками» с активированной Х-хромосомой, на которой расположен дефектный ген GLA , как описано выше. Этого переноса фермента достаточно для предотвращения уничтожения мутировавших клеток иммунной системой , но слишком мало, чтобы компенсировать генетический дефект, чтобы предотвратить накопление глоботриаозилцерамидов. По сравнению с другими лизосомальными ферментами поглощение α-галактозидазы A посредством переноса ферментов относительно низкое.
С помощью X-инактивации можно объяснить, что в среднем у гетерозиготных женщин заболевание становится симптоматическим намного позже и менее выражено, чем у мужчин. Однако этой модели недостаточно для понимания широкого спектра различных проявлений заболевания у женщин. Например, около 10% пациентов нуждаются в заместительной почечной терапии по мере прогрессирования заболевания , что соответствует «классическому фенотипу» у мужчин. С другой стороны, у других гетерозиготных женщин симптомы отсутствуют. Причина этого до сих пор полностью не выяснена.
Одна из гипотез предполагает, что нарушения в инактивации Х-хромосомы играют важную роль в диапазоне вариаций гетерозиготного фенотипа. Это известно как «кривая (англ. Skewed ) X-инактивация», при которой статистически ожидаемое соотношение 50: 50 между «мутантными» и «здоровыми» клетками значительно смещается. Этот сдвиг не вызван преимуществом роста мутировавших клеток. Гетерозиготные пациенты с фенотипом, при котором болезнь Фабри полностью развита, поскольку мутированная Х-хромосома активируется более чем в 95% клеток, являются признаком искривленной инактивации Х-хромосомы. Этому фенотипу соответствует примерно один из 200 пациентов с болезнью Фабри. Еще одним признаком искривленной Х-инактивации является гетерозиготная пара идентичных близнецов женского пола, в которой один из близнецов не имеет симптомов, а другой является клинически значимым. В исследовании с участием 28 пациентов склонная X-инактивация лейкоцитов была обнаружена у большинства пациентов , но это не имело никакого отношения к клиническим проявлениям заболевания или остаточной активности ферментов. Поэтому авторы не видят связи между фенотипом и изогнутой активацией X.
Варианты мутации
Дефекты гена GLA , вызванные мутациями , очень разнородны. На данный момент зарегистрировано более 500 различных мутаций. К ним относятся точечные мутации миссенс- и бессмысленного типов , мутации сплайсинга , небольшие делеции и вставки, а также большие делеции. Наиболее распространены точечные мутации (примерно 71%), за которыми следуют небольшие делеции и вставки, затрагивающие менее 60 нуклеотидов (примерно 27%), и большие делеции, затрагивающие один или несколько экзонов (примерно 2%). В большинстве случаев мутация приводит к полной потере активности фермента. Некоторые мутации, которые приводят к изменениям α-галактозидазы А и находятся достаточно далеко от активной области фермента, приводят только к небольшим структурным изменениям фермента, так что определенная остаточная активность фермента все еще присутствует. Такие мутации, как p.Met72Val, p.Gln279Glu или p.Met296Ile, характеризуются фенотипом легкой болезни. Продукты гена имеют нормальные значения для константы Михаэлиса K m и скорости оборота V max , но эти мутировавшие ферменты посттрансляционно деактивируются, а затем быстро разрушаются. Благодаря стабильности галактозы эти мутантные ферменты, очевидно, увеличиваются в лимфоцитах.
На гене GLA нет ярко выраженной « горячей точки» , которая особенно подвержена мутациям . Заметно увеличение количества перестроек ДНК в экзоне 7, который, очевидно, более подвержен перестройкам.
патология



Болезнь Фабри относится к группе, по крайней мере, из 50 членов лизосомных болезней накопления, а также к подгруппе сфинголипидозов . Заболевание основано на дефиците лизосомального фермента α-галактозидазы A. Этот дефицит вызывает накопление определенных продуктов метаболизма, таких как глоботриаозилцерамид (Gb3, Gl3, ранее также называвшийся церамид-тригексозидом), в эндотелиальных клетках различных систем органов. Снижение активности α-галактозидазы по существу приводит к накоплению глоботриаозилцерамида. Кроме того, накапливается дигалактозилцерамид, особенно в почках, и глоботриаозилшингозин (лизо-Gb3, лизо-Gl3). Эти сфинголипиды являются важными компонентами клеточной мембраны .
Точные взаимосвязи между сниженной или даже полностью отсутствующей активностью α-галактозидазы А и патологическими процессами в пораженных органах, которые в конечном итоге приводят к болезни Фабри, еще не выяснены. Разнообразие пораженных органов предполагает, что вторичные биохимические механизмы, в которых играют роль сфинголипиды, определяют течение заболевания.
Симптомы, описанные в следующей главе, такие как прогрессирующая хроническая почечная недостаточность, во многих публикациях приписываются накоплению глоботриаозилцерамида в лизосомах эндотелиальных клеток. Однако ряд клинических эффектов, особенно при заместительной ферментной терапии болезни Фабри, не укладывается в эту явно упрощенную модель. Например, у некоторых пациентов могут наблюдаться прогрессирующие осложнения, что свидетельствует об отсутствии прямой корреляции между Gb3 и клиническими проявлениями болезни Фабри. Наблюдение за тем, что у значительной части женщин- носителей мутации GLA развиваются симптомы, похожие на симптомы гемизиготных пациентов, даже несмотря на то, что у этих пациентов имеется значительное количество циркулирующего фермента, не соответствует упрощенной модели . Кроме того, накопление Gb3 в лизосомах гемизиготных пациентов начинается в раннем детстве или пренатально задолго до развития клинически значимых симптомов. Ни у гемизиготных, ни у гетерозиготных пациентов нет корреляции между степенью заболевания и уровнем Gb3 в плазме или моче.
Поскольку болезнь не проявляется в детстве даже у пациентов без активности α-галактозидазы A, предполагается, что накопление Gb3 не является непосредственной причиной болезни Фабри. В настоящее время предполагается, что глоботриаозилсфингозин - метаболит глоботриаозилцерамида - в конечном итоге является причиной патологического повреждения при болезни Фабри. По крайней мере, в повреждении клубочков , которое приводит к почечной недостаточности при болезни Фабри, лизо-Gb3 играет решающую роль. Лизо-Gb3 высвобождает TGF-β1 и ингибитор макрофагов CD74 . Последующий патомеханизм напоминает диабетическую нефропатию .
Клиническая картина


Формы и степень тяжести курса
Различают два типа пациентов и формы болезни Фабри: «классические» гемизиготные пациенты, у которых α-галактозидаза А не имеет активности, и «атипичные» гетерозиготные пациенты, у которых фермент все еще имеет остаточную активность. Классическое течение болезни проявляется в раннем появлении симптомов, как правило, в нескольких органах. Напротив, у атипичных гетерозиготных пациентов симптомы появляются намного позже. Кроме того, в таких случаях заболевание может быть локализовано, например, в сердечной мышце . У пациентов мужского пола с детства развиваются симптомы, характерные для болезни Фабри. У женщин это часто бывает только в возрасте от 40 до 50 лет. Из-за остаточной активности α-галактозидазы А симптомы часто менее выражены.
Симптомы сложны и могут проявляться по-разному от человека к человеку. Ранние симптомы очень важны для постановки диагноза. С другой стороны, самые поздние симптомы определяют смертность (уровень смертности) пациентов.
Индекс серьезности - это ключевой показатель , определяющий тяжесть заболевания. У пациентов с болезнью Фабри мужского пола пенетрантность была оценена как 100%, а степень тяжести - 84%. Для пациентов значения пенетрантности составляют 70%, а степени тяжести - 4%.
качество жизни
Симптомы болезни приводят к низкому качеству жизни, особенно у пациентов мужского пола. Это похоже на больных СПИДом. У пациентов с болезнью Фабри качество жизни находится на том же уровне, что и у пациентов с рассеянным склерозом или ревматоидным артритом .
Болезнь Фабри оказывает значительное негативное влияние на психосоциальную среду пострадавших. Более половины пациентов-мужчин не состоят в браке. Большая часть безработных. Депрессия чрезвычайно распространена у пациентов с болезнью Фабри. Они недостаточно диагностируются или получают недостаточное лечение и значительно снижают качество жизни пациентов. Согласно одному исследованию, 46% пациентов страдают депрессией, а 28% - депрессией, которая достаточно серьезна, чтобы иметь клиническое значение. Значения выше 26 достигаются по шкале Гамильтона . В отличие от нормального населения, доля мужчин с тяжелой депрессией (36%) выше, чем у женщин (22%).
В нескольких исследованиях рекомендуется психиатрическая и нейропсихологическая оценка пациентов с болезнью Фабри. В частности, пациенты жалуются на физические жалобы, грусть и эмоциональные переживания . Физические жалобы усиливаются при стрессе Психологические тесты показывают расстройства поведения выше среднего, недоверие, защитное отношение, эмоциональное возбуждение и чувство изоляции. Результаты этих тестов во многом схожи с результатами пациентов, страдающих болью.
Ранние признаки и симптомы


Боль
Одним из первых симптомов классической формы болезни Фабри является боль в руках и ногах, акре . Эти акропарестезии появляются в детстве. Они вызваны повреждением тонких нервных волокон ( невропатия мелких волокон ) вегетативной и периферической соматической нервной системы . От этой боли страдают от 60 до 80% мальчиков и девочек с классической формой.
Пациенты описывают два типа боли: периодически повторяющиеся приступы боли, также известные как «кризы Фабри», с жгучей болью, которая распространяется от рук и ног к другим частям тела, и хроническая боль, которая соответствует жжению и покалыванию. парестезии. Кризис Фабри может быть вызван лихорадкой, физическими упражнениями, стрессом, истощением и быстрыми перепадами температуры. Эти симптомы иногда неправильно интерпретируются как ревматические жалобы, синдром Рейно , системная красная волчанка и, прежде всего, как боли роста .
Боль обычно утихает в зрелом возрасте. У мальчиков они возникают раньше и чаще, чем у девочек. Мальчикам в среднем семь лет, а девочкам - девять лет. Боль оказывает значительное негативное влияние на качество жизни пациента.
Жалобы со стороны желудочно-кишечного тракта
Жалобы со стороны пищеварительного тракта - еще один частый, в основном недооцененный ранний симптом болезни Фабри. Эти недуги обычно сохраняются в зрелом возрасте. Пациенты жалуются на боли в животе , чаще всего после еды, диарею , тошноту и рвоту . Это, в свою очередь, может быть причиной анорексии (потери аппетита). Эти желудочно - кишечные симптомы, вероятно , вызваны Gb3 отложениями в вегетативных ганглиях (ганглии autonomica) из в кишечнике и брыжеечные кровеносных сосудами.
Ангидроз
Многие пациенты с болезнью Фабри не могут выделять пот ( ангидроз ) или могут делать это только в очень ограниченной степени (гипогидроз). Поэтому значения импеданса кожи сравнительно высоки. Ангидроз / гипогидроз может привести к непереносимости жары и значительным ограничениям в занятиях спортом у пострадавших. В исследовании с участием 714 пациентов с болезнью Фабри у 53% мужчин и 28% женщин был диагностирован ангидроз. Причиной снижения способности выделять потоотделение является накопление липидов в нейронах вегетативной нервной системы.
Ангиокератомы
Самый простой ранний симптом болезни Фабри - ангиокератома. Это доброкачественные красно-пурпурные изменения кожи с небольшими бугорками. Обычно они образуются на ягодицах , паху , пупке и бедрах . Иногда поражаются и слизистые оболочки , например, во рту. В большинстве случаев ангиокератомы представляют собой небольшие поверхностные ангиомы, вызванные повреждением эндотелия сосудов кожи в сочетании с расширением сосудов кожи. Они увеличиваются в количестве и размере с возрастом и могут происходить индивидуально или в группах. Помимо ангиом, причиной ангиокератомы также были случаи телеангиэктазии и подкожного отека .

Гистологический препарат образца кожи, взятого при биопсии у пациента с болезнью Фабри. На изображении, полученном с помощью светового микроскопа, типичные поражения кожи можно увидеть в виде небольших поверхностных ангиом.
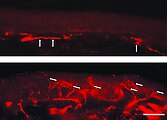
Флуоресцентный микроскоп изображения в замороженных срезов на коже пациента с болезнью Фабри.
Обращает на себя внимание отсутствие внутриэпидермальных нервных волокон и наличие волокон, принадлежащих субэпидермальному нервному сплетению (стрелки). С другой стороны, нижний образец кожи берется со спины пациента. Здесь заметна плотная иннервация эпидермиса (стрелки).
Рисунок после наблюдения под микроскопом карминного окрашивания эпидермиса с помощью ангиокератома, акварель из статьи Иоганна Фабри от 1898 года.



Вихревая кератопатия

Характерное помутнение роговицы - наиболее частый ранний симптом болезни Фабри. Они могут быть точно диагностированы с помощью щелевой лампы и встречаются почти у всех гемизиготных пациентов. Эта форма помутнения роговицы известна как вертициллезная роговица или вихревая кератопатия . Он встречается с обеих сторон и имеет характерный кремовый вихревой узор. Помутнение не влияет на остроту зрения. Некоторые препараты, такие как амиодарон и хлорохин, также вызывают вихревую кератопатию при длительном приеме.
Поздние симптомы
Повреждение почек

Как и большинство симптомов болезни Фабри, поражение почек прогрессирует. Он заканчивается терминальной почечной недостаточностью и приводит к значительному сокращению продолжительности жизни. В классической клинической картине болезни Фабри накопления Gb3 в эндотелиальных клетках клубочка , мезангиальных клетках , подоцитах и клетках интерстиция приводят к повреждению почек. Эти клетки представляют собой дифференцированные эпителиальные клетки . Скопления гликосфинголипидов также можно обнаружить в эпителии петли Генле и дистальных канальцах, а также в эндотелии и гладкомышечных клетках артериол почек. Эти отложения Gb3 в цитоплазме могут быть отчетливо видны в просвечивающем электронном микроскопе (ПЭМ). Они имеют форму миелиновых структур и упираются в ядро клетки . По мере увеличения накопления Gb3 мезангиум расширяется с последующим сегментарным или глобальным гломерулосклерозом с утолщением базальных мембран . В качестве возможных механизмов обсуждаются микрососудистые поражения и повреждение подоцитов, которые важны для работы фильтра, а также эпителиальные клетки канальцев.
При классическом течении болезни поражение почек начинается на втором-третьем десятилетии жизни . Прежде всего, может наблюдаться микроальбуминурия , то есть выведение небольшого количества протеина альбумина с мочой, которая перерастает в протеинурию (выведение большего количества протеина с мочой). Курс аналогичен диабетической нефропатии и напрямую способствует прогрессированию нефропатии Фабри. С возрастом протеинурия обостряется. По мере прогрессирования болезни развивается изостенурия , а это означает, что почки полностью теряют способность концентрироваться или разжижать. Это сопровождается изменениями канальцевой реабсорбции , секреции и экскреции .
Первоначально поражение почек маскируется клубочковой гиперфильтрацией . Но как только критическое количество нефронов повреждено, функция почек постепенно ухудшается. Скорость клубочковой фильтрации , показатель эффективности фильтрации почек, при отсутствии лечения снижается примерно на 12 мл / мин в год. На третьем-пятом десятилетии жизни функция почек постепенно ухудшается, и возникает почечная азотемия - аномальное увеличение содержания азотсодержащих продуктов метаболизма, таких как мочевина и креатинин, в крови. На этой стадии фиброз , склероз и атрофия канальцев преобладают в нефропатии Фабри и в конечном итоге приводят к терминальной почечной недостаточности, которая возникает у пациентов мужского пола в четвертом-пятом десятилетии. Около 17% всех мужчин и 1% всех пациентов с болезнью Фабри женского пола развивают терминальную стадию почечной недостаточности и нуждаются в диализе. Половина пациентов моложе 53 лет. Более чем у половины пациентов с болезнью Фабри в процессе болезни развивается нефропатия . Терминальная почечная недостаточность является основным фактором заболеваемости и смертности . Без заместительной почечной терапии уремия (отравление мочой) неизбежно приводит к смерти.
- Образцы тканей почек от пациентов с болезнью Фабри
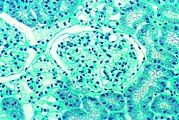
Эта световая микрофотография показывает накопление Gb3 в эндотелии клубочка, в мезангиальных клетках, клетках интерстиция и подоцитах.
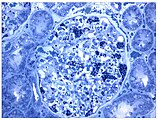
Также изображение светового микроскопа. Повышенное накопление Gb3 в подоцитах было видно по пурпурному окрашиванию.

Изображение ПЭМ показывает массивное электронно-плотное (= черное) скопление гликосфинголипидов в лизосомах подоцитов.

Также изображение ПЭМ. На нем видны включения гликосфинголипидов разной формы и размера в клетках дистального канальца.
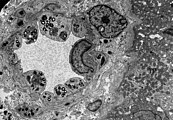
ПЭМ-изображение эндотелия и гладкомышечных клеток почечной артерии с включениями гликосфинголипидов





Повреждение сердца

Приблизительно от 40 до 60% пациентов с болезнью Фабри проявляют сердечные симптомы, такие как гипертрофия левого желудочка (ГЛЖ, утолщение сердечных стенок левого желудочка ), сердечная аритмия, стенокардия (приступообразная боль в груди) и одышка (затрудненное дыхание). ). Аритмия и нарушение вариабельности сердечного ритма являются вызваны по синусового узла , в проводящей системе , и нарушение баланса между симпатической и парасимпатической тон . Диастолическая дисфункция и гипертрофия левого желудочка являются основными симптомами болезни Фабри. Эти симптомы обычно более серьезны у мужчин, чем у женщин. Ишемия миокарда (нарушение кровообращения сердечной мышцы) является результатом плохого коронарного кровотока.
В пожилом возрасте прогрессивно развивается фиброз миокарда , который является как обратимо интерстициальным, так и необратимо поврежденным (замещающий фиброз) . Необратимые рубцовые фиброзы почти во всех случаях образуются сначала в задней боковой стенке сердца и в средней части миокарда. У пациентов в терминальной стадии трансмуральный (покрывающий всю толщину стенки сердца) рубцовый фиброз постепенно снижает сердечную функцию до точки застойной сердечной недостаточности . Злокачественные аритмии являются причиной большинства случаев сердечной смерти у пациентов с болезнью Фабри.
Структурные изменения левого желудочка сердца часто встречаются у пациентов с болезнью Фабри. В основном концентрические гипертрофии можно увидеть с помощью эхокардиографии (ультразвуковое исследование сердца) или магнитно-резонансной томографии сердца (МРТ). Поскольку задняя стенка левого желудочка сердца с возрастом становится тоньше из-за замещающего фиброза, измерение толщины перегородки - то есть толщины перегородки между левой и правой половинами сердца - особенно важно. Несмотря на структурные изменения, систола , фаза, в которой кровь вытесняется из левого и правого желудочка, по- видимому, в значительной степени сохраняется при измерении обычными методами. Кардиомиопатия, вызванная болезнью Фабри, характеризуется уменьшением сокращения и расслабления сердечной мышцы. Тканевый допплер ( визуализация скорости ткани и визуализация скорости деформации ) может количественно оценить функцию сердечной мышцы. С помощью этого метода кардиомиопатию можно диагностировать еще до того, как разовьется гипертрофия левого желудочка.
- Эхокардиографы пациентов с болезнью Фабри

Парастернальная длинная ось: четко видимая гипертрофия левого желудочка с увеличенной толщиной перегородки.

Парастернальная короткая ось: изображение также показывает гипертрофию левого желудочка.

Тканевая допплерэхокардиография митрального кольца (митрального кольца) с почти нормальной систолической функцией



У многих пациентов с болезнью Фабри правый желудочек также гипертрофирован (гипертрофия правого желудочка , RVH). Желудочек нормальных размеров, систола тоже в норме; диастолическая функция значительно снижена. Две трети пациентов с ГЛЖ также проявляют симптом РВГ. Гипертрофия правого желудочка - вероятная причина того, что пациенты с хорошей функцией левого желудочка сердца имеют плохую физическую выносливость и страдают органегалией (увеличенные органы) и лимфедемой .
Из-за нарушения функции сердца на электрокардиограммах (ЭКГ) взрослых пациентов с болезнью Фабри с классической формой заболевания наблюдаются характерные изменения.
- Электрокардиограммы (ЭКГ) пациентов с болезнью Фабри
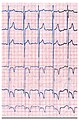
ЭКГ пациента с болезнью Фабри показывает гипертрофию левого желудочка с повышенным индексом Соколова-Лиона , уменьшенным сегментом ST и отрицательными зубцами Т в левых отведениях ЭКГ .

24-часовая ЭКГ часто проводится до и после заместительной ферментной терапии, если пациент сообщает об аномальном сердечном ритме или учащенном сердцебиении.


Цереброваскулярное повреждение
Ранние симптомы периферических невропатий , которые обычно появляются в подростковом возрасте, часто сопровождаются цереброваскулярными заболеваниями и вегетативными дисфункциями (заболеваниями или функциональными нарушениями вегетативной нервной системы). Некоторые из особенно неблагоприятных неврологических характеристик болезни Фабри вызваны церебральными многоочаговыми (мультифокальными) нарушениями кровообращения в мелких кровеносных сосудах. Цереброваскулярные изменения могут вызывать множество различных признаков и симптомов. Спектр варьируется от головных болей и головокружения до транзиторных ишемических атак и ишемических инсультов до сосудистой деменции .
Распространенность инфаркта головного мозга составляет около 6,9% у мужчин и 4,3% у женщин. Это намного выше, чем у населения в целом. Средний возраст первого инсульта составляет около 39 лет для мужчин с болезнью Фабри и 46 лет для женщин. Инфаркт мозга нередко бывает первым проявлением болезни Фабри. В большинстве случаев инфаркт мозга возникает из-за мелких кровеносных сосудов. Кроме того, в качестве спускового механизма были описаны долихоэктазии (син. Дилатационные артериопатии , расширение артерий) вертебробазилярного кровообращения. Образование тромбов , возможно , способствуют увеличению адгезии из нейтрофилов и моноцитов в эндотелий , или локально увеличение кровотока ( гиперперфузия ). Уровень фермента миелопероксидазы в сыворотке крови является биомаркером риска васкулопатии у мужчин с болезнью Фабри .
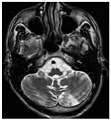
Осевая МРТ головного мозга 27-летнего пациента с болезнью Фабри с ишемическим инсультом показывает инсульт в левом полушарии мозжечка. В остальном у пациента отсутствовали симптомы заболевания.

Сагиттальная (A) и аксиальная (B) МРТ, взвешенная по T 1, показывает симметрично высокий сигнал в таламусе (так называемый пульвинарный признак ) 66-летнего пациента мужского пола. (C) и (D), также взвешенные по T 1 , показывают признак Pulvinar у 42-летнего пациента.

В время пролета магнитный резонанс ангиография четырех пациентов с болезнью Фабри показывает расширенную (экстатические) кровеносные сосуды (dolichoectasia из вертебробазилярного кровообращения).




Дальнейшие отдаленные последствия



Повреждение почек, сердца и мозга является основным фактором смертности от болезни Фабри. Другие долгосрочные эффекты клинически значимы, но не влияют на смертность от болезней или вносят незначительный вклад. Например, широко распространено поражение органов слуха и равновесия. 80% пациентов мужского пола и 77% женщин демонстрируют прогрессирующую потерю равновесия. Функцию органа равновесия можно проверить с помощью теста на импульс головы . У пациентов с гемизиготом и классическим течением заболевания прогрессирующая потеря слуха и внезапная глухота встречаются чрезвычайно часто. Накопление α-галактозидазы А также может привести к шуму в ушах и головокружению .
Дыхательные пути также поражаются этим заболеванием у многих пациентов с болезнью Фабри. Затрудненное дыхание ( одышка ), хронический кашель , а также нытье и хрипы характерны для обоих полов. Согласно исследованию, у 61% мужчин и 26% женщин имеется обструкция дыхательных путей .
Изменения скелета, которые существенно влияют на плотность костной ткани, также являются частым поздним симптомом болезни Фабри. В одном исследовании с помощью двойной рентгеновской абсорбциометрии (DXA) у 88% пациентов со средним возрастом 31 год и классическим течением заболевания была диагностирована либо остеопения, либо запущенная стадия, остеопороз . В последующем более крупном исследовании было обнаружено, что около 50% пациентов с болезнью Фабри страдают остеопенией. Пониженная плотность костной ткани может привести к самопроизвольным переломам .
диагноз
Важность ранней диагностики
Как можно раньше диагностировать болезнь Фабри важно по нескольким причинам. С одной стороны, возможность причинно-следственного лечения заболевания существует с 2001 года. Качество жизни пострадавших может быть значительно улучшено, а повреждение органов может быть, по крайней мере, уменьшено или отложено. С другой стороны, генетическая предрасположенность членов семьи к заболеванию может быть определена до появления первых симптомов. В таких случаях возможно наблюдение за развитием болезни и ранняя терапия до того, как болезнь станет симптоматической.
Неправильный диагноз
Из-за редкости болезни большинство педиатров и терапевтов ошибочно диагностируют болезнь Фабри и лечат ее соответствующим образом. В исследовании 2010 года были проанализированы истории болезни 45 пациентов с болезнью Фабри. Большинство пациентов в молодости жаловались на невропатическую боль как на первый симптом болезни - в большинстве случаев это ошибочно принимали за « ревматическую лихорадку » . Семь пациентов лечились пенициллином в течение многих лет . У десяти пациентов с болями в животе было диагностировано пищевое отравление или «неспецифическая боль». Первоначальный симптом ангидроза не мог быть отнесен к какой-либо причине, и ангиокератомы были интерпретированы как петехии . В среднем на постановку правильного диагноза болезни Фабри уходило 19,7 года. В предыдущем исследовании с 366 пациентами эта разница во времени составляла 13,7 года для мужчин и 16,3 года для женщин. Британское исследование 2001 года показало, что средний возраст пациентов мужского пола для первоначального диагноза составляет 22 года, что в среднем ставится через 8 лет после появления первых симптомов.
За долгий период между появлением первых симптомов и постановкой правильного диагноза многие пациенты прошли долгую и разочаровывающую поездку от врача к врачу. В большинстве случаев правильный диагноз ставится случайно у офтальмолога через вертициллезную роговицу (вихревая кератопатия) или у дерматолога через ангиокератому.
Правильный диагноз
При классической форме болезни Фабри клиническая картина может внести значительный вклад в ранний правильный диагноз; особенно ангиокератомы и вихревые кератопатии. У пациентов мужского пола определение активности α-галактозидазы A в плазме или лейкоцитах с помощью ферментного анализа обеспечивает диагностическую надежность . Определение из плазмы иногда может привести к неправильному диагнозу болезни Фабри, поэтому рекомендуется проверять результат с определением активности через лейкоциты. У пациенток этого метода часто бывает недостаточно. Он терпит неудачу более чем у 30% пациентов с болезнью Фабри, потому что их остаточная ферментная активность слишком высока для теста. Поэтому всем пациентам с подозрением на болезнь Фабри следует диагностировать генотипирование.
Ген секвенирован и сопоставлен с известными мутациями GLA. Кроме того, полезно определение биомаркера Lyso-Gb3. Если активность фермента неясна, значение лизо-Gb3 выше 1,3 нмоль / л может указывать на болезнь Фабри у людей с неспецифическими симптомами болезни Фабри (ГЛЖ или ХБП и т. Д.). Для измерения активности ферментов и лизо-Gb3, а также для генетического анализа доступен анализ сухой крови (Dried Blood Spot, DBS), который можно легко интегрировать в повседневную практику: для этого помещается несколько капель крови по карте сухой крови. После того, как они высохнут, карта отправляется по почте в специализированную лабораторию. Там кровь удаляется с фильтрующей карты и обрабатывается для дальнейших анализов. Lyso-GL-3 - хороший маркер для надежного исключения классической болезни Фабри. Людей с неопределенным значением GLA, у которых проявляются неспецифические симптомы болезни Фабри (ГЛЖ или ХБП и т. Д.) И отсутствуют характерные, фенотипические или биохимические признаки классической болезни Фабри, следует лечить с повышенными значениями лизо GL-3> 1,3 нмоль / л, думая о болезни Фабри. .
Уровни Gb3 в плазме и моче в принципе также можно использовать для диагностики. Значения мочи более надежны по своей информативности у пациентов мужского и женского пола, чем значения в плазме; однако у некоторых пациентов с поздней формой болезни Фабри или с определенными формами мутации (например, p.Asn215Ser) концентрация Gb3 в моче нормальная.
Синдром Кобба необходимо дифференцировать от дифференциального диагноза .
Пренатальная диагностика
Диагностика болезни Фабри возможна пренатально, т.е. пренатально . Это может включать биохимическую или молекулярную пренатальную диагностику . В первом случае активность α-галактозидазы A ворсинок хориона может быть измерена либо непосредственно, либо в культуре клеток . Удаление ворсин хориона возможно на 10 неделе беременности . Диагностика культивированных амниотических клеток (клеток околоплодных вод ), которые удаляются из околоплодных вод путем амниоцентеза , возможна примерно на 14 неделе беременности. Определение генотипа с помощью анализа ДНК (генетического теста) более сложное . Генетическое консультирование , как правило , проводятся до пренатальной диагностики. По этическим причинам пренатальная диагностика болезни Фабри, особенно плодов женского пола, является очень спорным вопросом. С появлением заместительной ферментной терапии это обсуждение распространилось на плод мужского пола. Некоторые авторы рекомендуют пренатальную диагностику только плодам мужского пола. Определение пола плода возможно на 9-11 неделе беременности по крови матери.
Диагноз предимплантационного в принципе возможен и уже проведен. Пока (по состоянию на сентябрь 2011 г.) публикаций по этому поводу нет.
Скрининг новорожденных
Болезнь Фабри в настоящее время не входит в число обследований новорожденных в Германии и Австрии. Предпосылка , что скрининг для конкретного врожденного порока только имеет смысл , если есть вариант лечения для него. С появлением заместительной ферментной терапии эта предпосылка при болезни Фабри устарела. Быстрый и относительно недорогой анализ некоторых лизосомных болезней накопления может быть проведен из сухих образцов крови ( сухих пятен крови , DBS) с помощью ВЭЖХ-МС . В настоящее время проводится несколько крупномасштабных исследований для проверки надежности процедуры. Plasma-Lyso-GL-3 может быть легко измерен с помощью анализа сухой крови и является эффективным и специфическим биомаркером для скрининга новорожденных с подозрением на болезнь Фабри до того, как станут заметны первые симптомы.
терапия

Разработка эффективных методов лечения, особенно активных ингредиентов, чрезвычайно затруднена при болезни Фабри, как и при всех лизосомных болезнях накопления. С одной стороны, из-за низкой заболеваемости имеется очень мало пациентов для проведения клинических исследований, а с другой стороны, требования в отношении безопасности лекарств при длительном приеме очень высоки. Лекарство следует принимать пожизненно и в идеале до появления первых симптомов, то есть практически здоровыми пациентами. Из-за редкости заболевания рынок разработанного препарата крайне мал. Таким образом, высокие затраты на разработку, обычно используемые в фармацевтической промышленности, распределяются между небольшим количеством пациентов, что приводит к очень высоким затратам на лечение в расчете на одного пациента.
До 2001 г. пациентов с болезнью Фабри можно было лечить только симптоматически или паллиативно . До этого момента лечение состояло в основном из избегания раздражителей, вызывающих боль, таких как стресс, физическая нагрузка, тепло, солнечный свет и резкие перепады температуры. Были обезболивающие в высоких дозах . Ангидрозу удалось противодействовать, увеличивая потребление жидкости в жаркую погоду и избегая физических нагрузок. Для облегчения желудочно-кишечных заболеваний использовалась диета с низким содержанием жиров и лекарства. Диеты для почек назначены при легкой протеинурии. Для предотвращения инсультов прописаны антикоагулянты. Терминальная почечная недостаточность лечилась - как и сегодня - заместительной почечной терапией (диализ или трансплантация почки).
Ферментная заместительная терапия
Заместительная ферментная терапия (ФЗТ) в настоящее время (по состоянию на сентябрь 2011 г.) является единственным вариантом причинного лечения ( причинной терапии ) болезни Фабри. Для пациентов с болезнью Фабри в Европейском Союзе для лечения болезни используются два активных вещества: агалсидаза альфа и агалсидаза бета. Оба являются биотехнологическими вариантами α-галактозидазы A. Агалсидаза бета доступна для пациентов в США с апреля 2003 года. Агалсидаза альфа еще не одобрена в США (по состоянию на сентябрь 2011 г.). Помимо 27 стран ЕС, он одобрен в общей сложности в 45 странах, включая Канаду, Японию, Бразилию и Китай (по состоянию на май 2011 г.).
Оба препарата созданы с помощью генной инженерии. В то время как в agalsidase альфа человеческой линии клеток фибробластов производить фермент, используют в agalsidase бета - яичнике из китайского хомячка ( клетки СНО ) , были использованы. Агалсидаза бета - химерный белок . Два фермента идентичны по своей аминокислотной последовательности, но немного отличаются по типу гликозилирования из-за разной формы экспрессии белка во время продукции . Основные различия заключаются в соотношении сиаловых кислот и маннозо-6-фосфата. Агалсидаза бета имеет более высокую долю полностью сиалилированных олигосахаридов и более высокую степень фосфорилирования. В тестах in vitro агалсидаза бета показала повышенное связывание с рецептором маннозо-6-фосфата и более высокое поглощение фибробластами Фабри. Напротив, никаких функциональных различий между двумя ферментами in vivo выявить невозможно. Они также неотличимы по степени перекрестной реактивности антител . Оба фермента необходимо вводить внутривенно. Ферменты, которые должны оказывать системное действие, обычно недоступны для перорального применения, поскольку они в значительной степени распадаются на свои компоненты (аминокислоты) в кишечнике. Агалсидаза альфа вводится в течение 40 минут в дозе 0,2 мг / кг массы тела каждые две недели . Агалсидаза бета вводится в том же ритме. Однако доза составляет 1 мг / кг массы тела, а время инфузии составляет первоначально четыре часа. Это время инфузии может быть сокращено до 90 минут при хорошей переносимости.
эффективность

Из-за медленного прогрессирования болезни Фабри на протяжении десятилетий, а также из-за ее редкости до сих пор имеется лишь несколько надежных данных о долгосрочном успехе лечения. В сравнительном исследовании в течение 24 месяцев не было обнаружено различий между агалсидазой альфа и агалсидазой бета по основным измеряемым параметрам заболевания. Заместительная ферментная терапия может значительно снизить накопление Gb3 в лизосомах, особенно в эндотелиальных клетках. Это имеет большое значение для лечения основных факторов заболеваемости (почечная недостаточность, сердечные и цереброваскулярные заболевания). Однако эффективность терапии у пациентов с запущенными симптомами довольно скромна. Поэтому как можно раньше лечение особенно важно для успеха терапии. Распад Gb3 при заместительной ферментной терапии зависит от типа клеток. В почках, помимо эндотелия сосудов, Gb3 также расщепляется в мезангиальных клетках клубочка и клетках интерстиция коркового вещества почек. Значительно хуже снижается Gb3 в гладких мышцах артериол и мелких артерий, подоцитах и в эпителии дистального канальца.
В клинических исследованиях было показано, что агалсидаза альфа уменьшает боль и гипертрофию левого желудочка. Стабилизировалась функция почек, улучшился слух и потоотделение. В целом качество жизни значительно повысилось. У пациентов с хронической почечной недостаточностью прогрессирование до терминальной почечной недостаточности может быть отложено. Во время лечения агалсидазой бета в различных клетках было продемонстрировано истощение Gb3. Повторного накопления Gb3 тоже нет. Введение агалсидазы бета значительно снижает риск серьезного клинического события (например, инфаркта миокарда, терминальной почечной недостаточности или смерти).
Побочные эффекты
Около половины всех пациентов имеют реакции, связанные с инфузией, от легкой до умеренной, пик которых наступает между пятой и восьмой инфузией. Кроме того, у некоторых пациентов наблюдается жар и озноб . Эти побочные эффекты кратковременны, несерьезны и поддаются консервативному лечению. После трех-пяти лет лечения только у 10-20% пациентов наблюдаются реакции, связанные с инфузией, и, очевидно, развивается толерантность к инфузии. Точная причина реакций, связанных с инфузией, до сих пор неизвестна. Предполагается образование специфических антител IgG к введенному ферменту. Сероконверсия IgG была обнаружена у 24% пациентов, получавших агалсидазу альфа, и у 88% пациентов, получавших агалсидазу бета . Образование антител может происходить у пациентов, у которых отсутствует остаточная активность α-галактозидазы. Для вашей иммунной системы α-галактозидаза «нова» и «странна». Образование антител против двух препаратов агалсидазы сразу снижает их эффективность. Однако многоцентровое исследование показало, что «чрезмерное распыление» антител соответствующими количествами фермента приводит к сохранению низких уровней маркера заболевания lyso-Gb3.
Конкурентная ситуация и производственные проблемы
В 2015 году объем продаж Shire Pharmaceuticals составил 326 миллионов долларов США, а Genzyme - 188 миллионов долларов США. Между двумя компаниями идет жесткая конкуренция на европейском рынке, в то время как в Соединенных Штатах одобрен только Fabrazyme . Genzyme потеряла долю на европейском рынке, потому что из-за проблем с доставкой пациенты были переведены с полной дозы Fabrazyme на более низкие дозы или Replagal. Когда возникла проблема с поставками Fabrazyme, большая часть американских товаров была направлена на экспорт в Европу . В результате пациенты в США получали либо более низкую дозу, либо вообще не получали лекарств для лечения, в то время как в Европе около 400 пациентов получали адекватную полную дозу.
Проблемы с доставкой впервые возникли в 2009 году из-за заражения импортированными калицивирусами (тип: Vesivirus 2117) в единственные в мире биореакторы для производства Fabrazyme в Олстоне ( Массачусетс ). Хотя вирус безвреден для человека, он оказал значительное влияние на урожайность. С другой стороны, это не повлияло на качество продукта. Производство также было остановлено на длительное время в связи с проведением дезинфекционных мероприятий. Genzyme была оштрафована на 175 миллионов долларов.
В августе 2010 года три пациента из США с болезнью Фабри обратились в Национальный институт здоровья (NIH) с петицией в пользу Genzyme с просьбой предоставить другим компаниям заместительную ферментную терапию на основе March-in Rights . Марш в области прав являются специальными правами вмешательства со стороны государства , которые существуют , когда научные исследования и разработки финансируются государством. Закон Бэя-Доула 1980 года позволяет университетам, среди прочего, самим продавать результаты финансируемых государством исследований. В свою очередь, Закон Бэя-Доула позволяет финансирующим государственным учреждениям отменять исключительные права на интеллектуальную собственность (в данном случае, патенты), например, когда люди подвергаются риску. Агалсидаза бета была разработана в Медицинской школе Маунт-Синай на средства NIH и защищена двумя патентами (5 356 804 и 5 580 757). Genzyme является эксклюзивным лицензиатом обоих патентов. Ходатайство пациента было отклонено NIH в декабре 2010 года. Основная причина, приведенная властями, заключается в том, что дальнейшее лицензирование третьим сторонам не решит проблемы, указанные заявителями - по сути, острое узкое место в поставках. С момента выдачи каждой лицензии до появления препарата от другого производителя потребуется несколько лет, поскольку необходимы комплексные клинические исследования и процессы утверждения. Кроме того, Genzyme обещает снова выйти на полную производственную мощность в первой половине 2010 года.
Проблемы с производством Fabrazyme сохранялись и в сентябре 2011 года. В первом квартале 2012 года производство началось на новом предприятии в Фрамингеме, штат Массачусетс , и предприятие было одобрено FDA. 100 пациентов из США в настоящее время (по состоянию на сентябрь 2011 г.) получают конкурирующий продукт Replagal в рамках клинического исследования, одобренного FDA .
После того, как регулирующие органы США дали добро на производство Fabrazyme на заводе в Фрамингеме, штат Массачусетс, компания Sanofi Genzyme Corp. начала производство. с доставкой. FDA и EMA ранее одобрили завод в Фрамингеме для производства Fabrazyme в январе 2012 года.
В марте 2012 года было объявлено, что Shire Pharmaceuticals отозвала свои нормативные документы в США для Replagal. Несмотря на рекомендацию FDA США и статус Fast Track, предоставленный FDA , он не был одобрен для рынка США.
Срок действия патента 5 356 804 Медицинской школы Маунт-Синай истек 27 сентября 2015 г. для рынка США и в августе 2016 г. для европейского рынка.
Затраты на терапию
Стоимость лечения в Германии составляет около 250 000 евро на пациента в год, независимо от того, используется ли агалсидаза альфа или бета. Ампула с 3,5 мг агалсидазы альфа при дозировке 0,2 мг на кг веса тела во Франции стоит 1685 евро. За ампулу с 35 мг агалсидазы бета с дозировкой 1 мг на кг массы тела взимается 3370 евро. Это приводит к идентичным годовым затратам на лечение в расчете на одного пациента, которые во Франции составляют 161 781 евро на пациента весом 70 кг (по состоянию на 2009 г.). Поскольку в настоящее время заместительная ферментная терапия является единственным вариантом терапии, вызывающим болезнь Фабри, затраты на нее возмещаются государственным медицинским страхованием в Германии и не покрываются из бюджета рецептов врача, выписывающего рецепты.
Сопутствующие терапии
Заместительная ферментная терапия довольно успешно снижает невропатическую боль, особенно у молодых пациентов. Однако во многих случаях все же показана сопутствующая обезболивающая . Для лечения невропатической боли при болезни Фабри, карбамазепина , возможно , в сочетании с прегабалина, как рекомендуется в качестве первого выбора . Опиоиды также можно использовать при невыносимых болевых приступах . При желудочно-кишечных заболеваниях рекомендуются такие препараты, как метоклопрамид , которые противодействуют двигательным нарушениям в верхних отделах желудочно-кишечного тракта (нарушения моторики).
Если наблюдается повышенная экскреция белка с мочой, что свидетельствует о повреждении почек , прогрессирование повреждения почек может быть замедлено дополнительным лечением ингибиторами АПФ или антагонистами AT1 , двумя родственными классами гипотензивных препаратов. Если сердце уже повреждено, ингибиторы АПФ могут снизить сопротивление артериальных сосудов и, следовательно, артериальное кровяное давление. Это также снижает преднагрузку и постнагрузку сердца в случае миокардиальной недостаточности и увеличивает сердечный выброс . Частоту сердечных сокращений и потребность в кислороде миокарда может быть снижена с помощью бета - блокаторы . Сердечную аритмию можно исправить , например, амиодароном . Кроме того, есть еще хирургические мероприятия, такие , как имплантация в кардиостимулятора , в коронарных стентов , с искусственного сердечного клапана или аортокоронарное шунтирование .
Возможные варианты терапии в будущем
Во многих случаях заместительная ферментная терапия может остановить или, по крайней мере, уменьшить прогрессирование болезни Фабри. Лечение носит чисто паллиативный характер , а это означает, что полное излечение с его помощью невозможно. Кроме того, не все пациенты оптимально реагируют на этот вид лечения. Лекарственная форма определяется для внутривенного введения . Пероральный прием, который обычно предпочитают пациенты, невозможен. Во время всасывания в желудочно-кишечном тракте α-галактозидаза А расщепляется на неэффективные фрагменты. Поэтому одним из направлений разработки являются доступные для перорального применения лекарственные средства, которые по меньшей мере эквивалентны заместительной ферментной терапии с точки зрения их эффективности или могут дополнять их терапевтически.
Дальнейшее развитие получает и сама заместительная ферментная терапия. Возможный препарат будущего - это модифицированная форма фермента α-N-ацетилгалактозаминидазы (NAGA). α-N-ацетилгалактозаминидаза имеет структуру, очень похожую на α-галактозидазу A. В активном кармане они различаются только двумя аминокислотными положениями. Посредством целенаправленных изменений ( белковая инженерия ) в активном кармане этого человеческого фермента он может принимать Gb3 в качестве субстрата и расщеплять его, как α-галактозидаза А. Большим преимуществом модифицированной α-N-ацетилгалактозаминидазы является то, что она не обладает иммуногенностью у пациентов с болезнью Фабри . Ваша иммунная система «знает» α-N-ацетилгалактозаминидазу.
Шаперонная терапия

Большинство мутаций в гене GLA относятся к миссенс- типу . Генные продукты включают большое количество вариантов ферментов, которые в очищенной форме сравнимы по своей активности с α-галактозидазой А дикого типа , но имеют более низкую термическую и pH-стабильность. Эти мутантные варианты альфа-галактозидазы А представляет собой - как и все вновь синтезированных белков - в эндоплазматический ретикулум (ER) о «контроля качества» хроматографии ( белок Контроль качества , оборудование контроля качества белка ). Это гарантирует, что только правильно свернутые и модифицированные белки достигают своего места назначения. Белки, не прошедшие этот контроль качества, транспортируются из эндоплазматического ретикулума в цитозоль и расщепляются в протеасоме . Этот процесс называется ER-ассоциированной деградацией белка (ERAD, Endoplasmic Reticulum Associated Protein Degradation ). Неправильно свернутые варианты α-галактозидазы A из-за мутации отделяются и разбиваются в рамках контроля качества.
Однако этот процесс дает возможность терапевтического вмешательства. С помощью фармакологических шаперонов или химических шаперонов неправильную укладку мутировавших ферментов можно исправить. Фармакологические шапероны представляют собой небольшие молекулы ( маленькие молекулы ), которые служат для свертывания шаблона ферменту. Это сдвигает динамику укладки белка в сторону правильной конформации и стабилизирует ее. При правильной третичной структуре контроль качества в ER «пройден». Стабильный шаперон-белковый комплекс переносится через везикулы эндоплазматического ретикулума в аппарат Гольджи, а затем в лизосомы. Там фармакологический шаперон заменяется естественным субстратом (Gb3). Диссоциации комплекса шаперон-белок способствует высокая концентрация Gb3 и низкое значение pH в лизосоме.
Имин сахар 1-Deoxygalactonojirimycin (DGJ), международное неправительственное фирменное наименование Migalastat , является примером фармакологического компаньонки. Это аналог концевой галактозы Gb3 и обратимый ингибитор α-галактозидазы A. В большом количестве доклинических экспериментов было показано, что мигаластат способен повышать активность мутантных вариантов α-галактозидазы A. Это позволило, например, значительно снизить накопление Gb3 у мышей Fabry. Как небольшая молекула, мигаластат имеет очень широкое биораспределение в организме и может, например, достигать центральной нервной системы и преодолевать гематоэнцефалический барьер . Кроме того, он доступен перорально.
Мигаластат одобрен в Европейском Союзе с 2016 года. До сих пор неясно, какую долю пациентов с болезнью Фабри можно будет лечить этой формой терапии в будущем, поскольку не все из более чем 500 мутационных форм α-галактозидазы A, известных на сегодняшний день, можно активировать или правильно развить с помощью фармакологических шаперонов. При исследовании 299 форм мутаций 40 из них классифицированы как потенциально поддающиеся лечению фармакологическими шаперонами. Шансы на успех хорошие, особенно с мутациями, вызывающими неклассическое течение болезни. Перед возможным началом терапии обязательно будет анализ генотипа .
Субстрат-редукционная терапия

В то время как заместительная ферментная терапия при болезни Фабри пытается заменить отсутствующую или дефектную α-галактозидазу путем введения эффективного искусственно производимого фермента, чтобы иметь возможность расщеплять субстрат Gb3 в клетках, терапия субстрат-восстановлением (SRT) использует другой подход. Здесь пытаются уменьшить количество субстрата и тем самым предотвратить накопление Gb3 в клетках. Это возможно, например, путем ингибирования фермента глюкозилцерамидсинтазы (GCS, также называемой церамид глюкозилтрансферазой ). GCS катализирует первую стадию синтеза гликосфинголипидов и тем самым синтез последующих молекул, включая Gb3. Основная эффективность этого терапевтического подхода может быть продемонстрирована на модельном организме мышей. СРТ может стать в будущем вариантом дополнительного лечения болезни Фабри по сравнению с заместительной ферментной терапией. Примером ингибитора гликозилтрансферазы является миглустат . Этот препарат одобрен для лечения двух лизосомных болезней накопления: болезни Ниманна-Пика типа C и болезни Гоше типа 1. Однако в последнем случае только у пациентов, для которых ФЗТ не является вариантом лечения. Миглустат не одобрен для лечения болезни Фабри. Профиль побочных эффектов миглустата с диареей и периферической невропатией неблагоприятен для болезни Фабри и может еще больше усугубить симптомы заболевания. В случае болезни Фабри, при которой у большинства пациентов не наблюдается остаточной активности мутировавшего фермента, применение SRT в качестве монотерапии маловероятно. Поэтому будущие подходы к СРТ больше нацелены на поддержку заместительной ферментной терапии. Одно из потенциальных лекарств - элиглустат .
Пересадка костного мозга
В модельном организме мышей с выключенным геном gal можно было продемонстрировать, что активность α-лактозидазы A увеличивается при трансплантации стволовых клеток из костного мозга мышей дикого типа . Это может быть достигнуто за счет снижения кондиционирования , для которого, очевидно, достаточно генной коррекции около 30%. Процедура и кондиционирование несут значительно более высокий риск, чем заместительная ферментная терапия. Нет никаких результатов или опыта использования этой формы терапии болезни Фабри.
Генная терапия
Генная терапия - это многообещающий вариант лечения в будущем, в котором есть надежда, что излечение может быть достигнуто с помощью одного лечения . ДНК или РНК с генетическим кодом гена GLA будут вставлены в клетки тела. Терапевтический подход будет заключаться в мутировании гена в причинной цепи → дефектный фермент → дефектная функция → отложение Gb3 → болезнь Фабри на одну стадию раньше, чем при заместительной ферментной терапии. В доклинических экспериментах на мышах Fabry были получены многообещающие результаты с различными векторами , особенно вирусными векторами . Пока невозможно предсказать, станет ли этот метод лечения доступным для пациентов с болезнью Фабри и когда это произойдет. В целом высокие ожидания в отношении генной терапии, которые существовали прежде всего на рубеже тысячелетий, в последние годы значительно снизились. При некоторых вариантах процесса, таких как использование ретровирусов для трансдукции , существует риск индуцирования рака за счет инактивации генов-супрессоров опухоли . Успешная соматическая генная терапия могла бы полностью излечить пролеченного пациента с болезнью Фабри, но дефектный ген GLA все еще оставался бы в его половых клетках . Таким образом, его дети по-прежнему имеют 50% шанс заболеть этой болезнью. Терапия зародышевой линии у людей, которая предотвратит передачу дефектного гена потомству, запрещена в Германии и большинстве других стран.
Модели животных
Генетически модифицированные организмы используются для разработки новых терапевтических возможностей при болезни Фабри и для исследования болезни. До сих пор не было обнаружено ни одной крупной животной модели болезни Фабри, поэтому в качестве модельного организма по существу используется мышь с нокаутом по a-gal-A (мышь a-gal-A - / 0 или «мышь Fabry»). В этом нокаутном мышее , гал ген выключен с помощью гена нокаута . Однако по сравнению с генетическим дефектом у людей это мало влияет на фенотип. Даже в возрасте 80 недель мыши клинически нормальны, имеют нормальные показатели крови и мочи и имеют такую же продолжительность жизни, как и мыши дикого типа . Гистопатологически можно продемонстрировать накопление Gb3 в печени и почках, что у старых мышей генотипа a-Gal A - / 0 вызывает морфологические изменения в ткани, но не приводит к каким-либо повреждениям. Доступность мышей Фабри сыграла важную роль в разработке заместительной ферментной терапии болезни Фабри, особенно на доклинической стадии . Мышей Α-Gal-A - / 0 также используют в доклинических исследованиях генной, шаперонной и субстратной терапии.
прогноз
С возрастом повреждение жизненно важных органов, вызванное накоплением Gb3, продолжает увеличиваться, пока эти органы полностью не утратят свою функцию. Терминальная почечная недостаточность и опасные для жизни сердечно-сосудистые или цереброваскулярные осложнения ограничивают продолжительность жизни нелеченных пациентов в среднем примерно до 50 лет, а пациентов - примерно до 70 лет. По сравнению с населением в целом это соответствует сокращению на 20-15 лет.
В одном исследовании основными причинами смерти были почечная недостаточность, цереброваскулярные заболевания (например, кровоизлияние в мозг или инсульт) и болезни сердца. Доля смертей от почечной недостаточности снизилась с введением диализа и трансплантации. Основной причиной смерти в период исследования с 2001 по 2007 годы были болезни сердца; 34% мужчин и 57% женщин. Причина изменения причин смерти видна в улучшении клинической помощи пациентам с терминальной стадией почечной недостаточности.
Пока нет статистически значимых данных о том, может ли заместительная ферментная терапия увеличить продолжительность жизни пациентов, и если да, то в какой степени.

Показатель выживаемости Каплана-Мейера для пациентов с болезнью Фабри мужского пола по сравнению с нормальной мужской популяцией. Средняя продолжительность жизни составляет 50 лет, что соответствует сокращению примерно на 20 лет. Данные относятся к 2001 году, когда заместительная ферментная терапия еще не была доступна.

Соответствующая выживаемость пациентов женского пола. Средняя продолжительность жизни составляет 70 лет, что соответствует сокращению примерно на 15 лет. Эти данные также за 2001 год.


История болезни
Болезнь Фабри была обнаружена относительно поздно как самостоятельный синдром. Первые публикации о болезни написаны Иоганном Фабри и Уильямом Андерсоном (оба в 1898 году). Общая средняя продолжительность жизни составляла около 30 лет в 1820 году и около 40 лет в 1900 году. Для сравнения, средняя продолжительность жизни пациентов мужского пола с болезнью Фабри сегодня составляет от 45 до 50 лет. Таким образом, болезнь встречается не только крайне редко, но и клинически незаметна, учитывая в целом короткую продолжительность жизни. В конце 18 века дерматологи могли описывать только кожные заболевания. Значение сопутствующих симптомов и взаимосвязь между ними были в значительной степени неизвестны.
Фаза эмпирической дерматологии не закончилась до 1970-х годов, когда появились микробиологические и иммунологические процедуры.
Иоганнес Фабри и его пациент

Немецкий дерматолог Йоханнес Фабри в то время работал в городской больнице Дортмунда . В декабре 1898 года он опубликовал в архиве дерматологии и сифилиса статью «Вклад в изучение узловатой пурпуры геморрагической» (Purpura papulosa haemorrhagica Hebrae) .
В своей статье Фабри назвал кожные поражения, вызванные ангиокератомой, Purpura papulosa haemorrhagica Hebrae . Фабри выбрал это имя,
«... потому что, прежде всего, мы должны избегать добавления новой клинической картины к большой и очень спорной группе лишайников; но прежде всего потому, что древнееврейский термин в слове «пурпура» также включает в себя пол и эпитет клиническое разнообразие и особенности узловой формы и папулезной сыпи ».
Фабри знал первое описание. Он написал: «Но сначала точный медицинский отчет об интересном и, как мне кажется, в литературе без аналогового случая».

Затем он описал историю болезни Эмиля Хонке, 13-летнего мальчика из Лангендрера . Его родители заметили небольшие узелки в подколенной области его колена, когда ему было девять лет. С годами сыпь распространилась по задней поверхности бедер до туловища. В двенадцать лет узелки появились и на впадине левого колена. В первые несколько лет никаких дальнейших симптомов не было, но затем у Эмиля становилось все слабее и падал аппетит. Само состояние кожи не доставляло ему дискомфорта - ни зуда, ни покалывания, ни боли, ни дискомфорта. Оба родителя, которым было за 40, описали Фабри как здорового. Дед по отцовской линии умер от почечной недостаточности в возрасте 49 лет. Фабри сделал выводы 15 апреля 1897 года. Он продержал Эмиля несколько дней в кожном отделении городской больницы Дортмунда и взял образцы тканей кожи. Фабри прописал ему железные капли и воздержался от местного лечения. Фабри описал узелки как от темно-синего до черного. Фабри описал ощущение прикосновения при поглаживании кожи груди как «типичное ощущение терки», подобное, например, болезни Дарье .
Перед публикацией Фабри представил гистопатологические данные, полученные от Эмиля Хонке в 1897 году, на встрече врачей в административном районе Арнсберг и в дерматологической секции собрания естественных исследователей Брауншвейга.

Спустя 17 лет, 11 февраля 1915 года, Эмиль Хонке вернулся в Фабри из-за своей болезни. Тем временем он полностью потерял из виду дело. Хонке тем временем работал маляром, а затем в течение трех лет работал шахтером.
«Мы полностью потеряли из виду этот случай и, конечно, были очень удивлены, когда в этом году [пациент] снова явился на обследование. Из анамнеза следует подчеркнуть, что состояние кожи не мешало ему заниматься своей профессией ».
В статье 1916 года об ангиокератомах Фабри опубликовал результат теста с двумя фотографиями своего пациента. Это показывает прогрессирующее распространение ангиокератом, особенно на спине и руках, по сравнению с изображением 1898 года. Фабри смог обнаружить в моче альбумин, то есть протеинурию. Однако он больше не обращал внимания на это открытие. Фабри прогнозировал, что из-за предшествующего течения болезни не следует ожидать угрожающего развития. Общее самочувствие не нарушается и вряд ли изменится в обозримом будущем. Он также не ожидал выздоровления, поскольку с годами кожные изменения постепенно усиливались. Фабри воздерживался от лечения, тем более что его пациент тоже не хотел этого. Однако он хотел бы попробовать лечение рентгеновскими лучами и радием .
В этой публикации Фабри предположил, что его случай универсальной ангиокератомы не имеет аналогов в литературе: «Единственный возможный случай, Андерсона, не ангиокератома, это множественные ангиомы». В этом пункте Фабри ошибался.
В 1930 году, в год своей смерти, Фабри опубликовал статью о наивиформной ангиокератоме . В нем он вернулся к случаю своего пациента. Эмиль Хонке умер в 1928 году в возрасте 43 лет от неуточненной болезни легких, но не от туберкулеза .
Уильям Андерсон и его пациент


В апреле 1898 года британский анатом Уильям Андерсон опубликовал пятистраничный отчет о пациенте с ангиокератомой (случай «Анджио-кератомы») в British Journal of Dermatology .
Уильям Андерсон впервые увидел своего пациента (WH) в декабре 1897 года. Ему было 39 лет, и он был художником по профессии. Его кожные изменения начались в возрасте одиннадцати лет; сначала на колени, затем на туловище и конечности. В 17 лет был достигнут максимальный спред. Андерсон зафиксировал распространение диффузной ангиокератомы в зарисовке, которая была частью его публикации . Семейный анамнез выявил аналогичные кожные изменения у матери, сестры и троих из четырех детей. При первом обследовании Андерсон обнаружил в моче пациента немного альбумина. После этого моча была нормальной. Через несколько дней в больнице очаги поражения несколько уменьшились. По просьбе пациента лечение не проводилось.
Андерсон предположил, что заболевание встречается у обоих полов и сопровождается обморожением или асфиксией . Он сказал, что во многих случаях могут быть затронуты члены семьи, но поскольку на момент анамнеза в семье его пациента не было известно о других случаях ангиокератомы корпорис диффузум , он не видел убедительных доказательств наличия наследственного заболевания.
WH умер в 1911 году от кахексии, вызванной туберкулезным энтеритом . Дочь и двое внуков также заболели болезнью Фабри.
Дальнейшие исследования болезни
В последующий период болезнь Фабри первоначально называлась Angiokeratoma corporis diffusum . Термин ангиокератома восходит к Эрнесту Виндхему Коттлу (? –1919), который в 1877 году впервые описал случай ангиокератомы у восьми пациентов с бородавчатыми наростами . Со временем нынешнее название болезни Фабри стало обычным явлением , хотя болезнь Андерсона-Фабри была бы более подходящей, учитывая исторический фон.
В 1925 году Иоганнес Вайксель (1882–?) Первым распознал характерные изменения сетчатки и конъюнктивы как симптом болезни Фабри. В 1947 году было обнаружено, что пара братьев впервые участвовала в прогрессировании болезни Фабри. Во время вскрытия рабочая группа дерматолога из Гронингена Максимилиана Руйтера (1900–1974) обнаружила аномальные вакуоли в кровеносных сосудах. Они первыми высказали подозрение, что болезнь Фабри - это систематизированная болезнь накопления. Поэтому в некоторых работах болезнь Фабри также называют синдромом Руйтера-Помпена-Вейерса.
Немецкий патолог Карл Скриба (1907–1978) из Университетского медицинского центра Гамбург-Эппендорф обнаружил липиды в вакуолях в 1950 году. Три года спустя вместе с Хансом Хорнбостелем он впервые смог обнаружить эндотелиальные жировые отложения у живого пациента с болезнью Фабри.
В 1958 году впервые был описан случай болезни Фабри у женщины.
Чарльз С. Суили и Бернард Клионски из Университета Питтсбурга охарактеризовали жировые отложения в почках 28-летнего пациента Фабри, умершего в 1963 году, и определили глоботриаозилцерамид. Они классифицировали болезнь как сфинголипидоз.
Тот факт, что болезнь имеет Х-сцепленное наследование, был доказан в 1965 году исследовательской группой, возглавляемой Джоном Опитцем из Университета Висконсина в Мэдисоне, с помощью анализа генеалогического древа . В том же году японский дерматолог Кен Хашимото , который живет в Соединенных Штатах, обнаружил тельца включения в клетках лизосом на изображениях ТЕМ образцов кожи пациентов с болезнью Фабри. Он первым заподозрил, что причиной болезни является дефект лизосомального фермента.
Разработка заместительной ферментной терапии
В 1967 году исследовательская группа из Национального института неврологических заболеваний и слепоты обнаружила, что дефицит церамидтригексозидазы является генетической причиной болезни Фабри. В 1970 году был идентифицирован фермент α-галактозидаза, два варианта которого A и B были выделены из плаценты человека в 1978 году ; дефицит варианта А ответственен за развитие болезни Фабри. Благодаря этим открытиям, сделанным в 1973 году, носителей признаков - также пренатально - можно было определить биохимически на основе активности ферментов.
Это положило начало заместительной ферментной терапии. Первоначально было начато комплексное выделение фермента из плаценты, селезенки и плазмы человека, а после инфузии α-галактозидазы А было обнаружено снижение глоботриаозилцерамида в крови пациентов с болезнью Фабри.
Достижения генной инженерии сделали возможным создание генетически модифицированных организмов . Междисциплинарная рабочая группа под эгидой Национального института здравоохранения в 1996 году преуспела в выведении гена α-Gal-A в модельном организме цветной мыши , создав первую «мышь Фабри». С развитием секвенирования ДНК были достигнуты дальнейшие успехи в исследованиях болезни Фабри. Знание последовательности гена GLA впервые позволило локализовать и охарактеризовать мутации и получить рекомбинантную ДНК .
С введением рекомбинантной ДНК в бактерии стало возможным продуцировать α-галактозидазу А как рекомбинантный белок и, таким образом, использовать его в достаточных количествах для доклинических и клинических исследований. Первые доклинические исследования были проведены в 1996 г. на мышах Fabry. В рамках клинического исследования в 1999 г. пациенты впервые получили генно-инженерную α-галактозидазу А. В начале августа 2001 г. агалсидаза бета (Genzyme Corporation) и агалсидаза альфа ( Transkaryotic Therapies , теперь Shire Pharmaceuticals) были одобрены в в то же время в Законе Европейского союза о лекарственных препаратах для лечения болезни Фабри. Fabrazyme был запущен 15 августа 2001 года, а Replagal - в Германии в середине октября того же года .
литература
Справочная литература
- Д. Эльштейн, Г. Альтареску, М. Бек (ред.): Болезнь Фабри. Verlag Springer, 2010, ISBN 978-90-481-9032-4 ограниченный предварительный просмотр в поиске книг Google
- Дж. А. Баррангер, М. А. Кабрера-Салазар (ред.): Лизосомные нарушения накопления. Verlag Springer, 2007, ISBN 978-0-387-70908-6 , ограниченный предварительный просмотр в поиске книг Google
- К. Ваннер: Болезнь Фабри: клиника, диагностика и терапия. Verlag Uni-Med, 2004, ISBN 3-89599-769-2 .
- А. Мехта, М. Бек, Г. Сандер-Плассманн (ред.): Болезнь Фабри. Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290683 (полный свободный доступ)
Техническая статья
- Б. Хоффманн, Э. Маятепек: Болезнь Фабри - часто наблюдается, редко распознается . В: Dtsch Arztebl Int. 106, 2009, с. 72-81. PMID 19623315
- Б. Гофманн: Болезнь Фабри: последние достижения в патологии, диагностике, лечении и мониторинге. В: Журнал редких заболеваний Orphanet. 4, 2009, 21. doi: 10.1186 / 1750-1172-4-21 , PMC 2768700 (полный текст), PMID 19818152 (обзорная статья в открытом доступе )
- К. Уайбра, М. Рис, К. Кампманн: Клинические проявления болезни Фабри у детей. (PDF; 2,8 МБ) В: Нейропедиатрия. 2, 2005, с. 44-48.
Научно-популярная и научно-популярная литература
- Джеральд Улиг - Ромеро: И все же я жив. Моя борьба с загадочной болезнью. Deutsche Verlags-Anstalt, 2009, ISBN 978-3-421-04326-9 .
- У. Шмидт: Редкое наследственное заболевание. (PDF; 6,7 МБ) В: JOGU. 186, 2003, с. 14-15.
- М. Павлик: Дайте мне место без страха! В: FAZ , 22 июня 2009 г. (рецензия на книгу Джеральда Улига)
- В. Хакенброх: жир в органе . В: Der Spiegel . Нет. 28 , 2006, стр. 111 ( онлайн ).
- Н. Шойфлер: Сирота медицины - Наследственная болезнь Болезнь Фабри не известна даже многим врачам. В: ФАЗ , 12 июня 2002 г., с. 10.
веб ссылки
- Болезнь Фабри. В: Интернет-Менделирующее наследование в человеке . (Английский)
- Болезнь Фабри. В: Orphanet (База данных редких заболеваний).
- Патология болезни Фабри - база данных патопических изображений Базельского университета ( PathoPic - инструкции (PDF); PDF; 2,2 МБ)
- Группа самопомощи при болезни Фабри e. В.
- Стефани Барбара Шац: Х-инактивация у гетерозиготных пациентов в отношении Х-сцепленной болезни Фабри. (PDF) Диссертация, Университет Людвига Максимилиана, Мюнхен, 2012 г.
Индивидуальные доказательства
- ↑ BJ Poorthuis, RA Wevers u. A .: Частота лизосомных болезней накопления в Нидерландах. В кн . : Генетика человека. Том 105, номера 1-2, июль-август 1999 г., стр. 151-156, PMID 10480370 .
- ^ PJ Meikle, JJ Hopwood et al.: Распространенность лизосомных нарушений накопления. В: JAMA. Том 281, номер 3, январь 1999 г., стр. 249–254, DOI : 10.1001 / jama.281.3.249 , PMID 9918480 .
- ↑ a b RJ Desnick, YAIoannou, CM Eng: Дефицит a-галактозидазы A: болезнь Фабри. В: CR Scriver , AL Beaudet, WS Sly, D. Valle (Eds.): Метаболические и молекулярные основы наследственных заболеваний. 8-е издание, McGraw-Hill Verlag, 2001, ISBN 0-07-913035-6 , стр. 3733-3774.
- ↑ Д. Марсден, Х. Леви: Скрининг новорожденных на лизосомные нарушения накопления. В кн . : Клиническая химия. Том 56, номер 7, июль 2010 г., стр. 1071-1079, DOI: 10.1373 / Clinchem.2009.141622 . PMID 20489136 . (Рассмотрение).
- ↑ М. Спада, С. Пальярдини и др.: Высокая частота поздних форм болезни Фабри, выявленная при скрининге новорожденных. В: Американский журнал генетики человека. Том 79, номер 1, июль 2006 г., стр. 31-40, DOI: 10.1086 / 504601 . PMID 16773563 . PMC 1474133 (полный текст).
- ↑ a b WL Hwu, YH Chien et al.: Скрининг новорожденных на болезнь Фабри на Тайване выявляет высокую частоту более поздней мутации GLA c.936 + 919G> A (IVS4 + 919G> A). В: Мутация человека. Том 30, номер 10, октябрь 2009 г., стр. 1397-1405, DOI: 10.1002 / humu.21074 . PMID 19621417 . PMC 2769558 (полный текст).
- ↑ б с д е е г ч я J к л м п о р Q R сек т у V ш х у г аа DP Germain: Болезнь Фабри. В: Журнал редких заболеваний Orphanet. Том 5, 2010 г., стр.30, DOI: 10.1186 / 1750-1172-5-30 . PMID 21092187 . PMC 3009617 (полный текст). (Обзор в открытом доступе ).
- ↑ Центр компетенции по болезни Фабри представляет себя. (PDF) ( Страница больше не доступна , поиск в веб-архивах ) Информация: ссылка была автоматически помечена как дефектная. Пожалуйста, проверьте ссылку в соответствии с инструкциями, а затем удалите это уведомление. (PDF; 595 kB) Госпиталь Кельнского университета, по состоянию на 1 сентября 2011 г.
- ↑ а б К. Веттер: Повторение: болезнь Фабри. (PDF) ( Страница больше не доступна , поиск в веб-архивах ) Информация: ссылка была автоматически помечена как дефектная. Пожалуйста, проверьте ссылку в соответствии с инструкциями, а затем удалите это уведомление. В: ЗМ. 101, № 1А, от 1 января 2011 г., стр. 41-45.
- ↑ a b KF Gold, GM Pastores et al.: Качество жизни пациентов с болезнью Фабри. В: Qual Life Res. Volume 11, Number 4, June 2002, pp. 317-327, PMID 12086117 .
- ^ HY Lin, KW Chong et al.: Высокая частота сердечного варианта болезни Фабри, выявленная при скрининге новорожденных в китайском населении Тайваня. ( Memento из в оригинале с 27 декабря 2016 года в интернет - архив ) Info: архив ссылка автоматически вставляется и еще не проверен. Проверьте исходную и архивную ссылку в соответствии с инструкциями, а затем удалите это уведомление. В кн . : Тираж. Сердечно-сосудистая генетика. Том 2, номер 5, октябрь 2009 г., стр. 450-456, DOI : 10.1161 / CIRCGENETICS.109.862920 . PMID 20031620 .
- ↑ П. Гаспар, Дж. Эррера и др.: Частота болезни Фабри у мужчин и женщин, находящихся на гемодиализе в Испании. В кн . : BMC Medical Genetics. Том 11, 2010 г., стр. 19, DOI: 10.1186 / 1471-2350-11-19 . PMID 20122163 . PMC 2837018 (полный текст).
- ↑ a b c d А. П. Бурлина, К. Б. Симс и др.: Ранняя диагностика вовлечения периферической нервной системы в болезнь Фабри и лечение нейропатической боли: отчет экспертной группы. В кн . : БМК неврология. Том 11, 2011 г., стр. 61, DOI: 10.1186 / 1471-2377-11-61 . PMID 21619592 . PMC 3126707 (полный текст).
- ^ A b М. Павелка, Дж. Рот: Функциональная ультраструктура: Атлас биологии и патологии тканей. Verlag Springer, 2005, ISBN 3-211-83563-6 , стр. 104-105, doi : 10.1007 / 3-211-30826-1_56 ограниченный предварительный просмотр в поиске книг Google
- ↑ omim.org: OMIM Gene / Loci: 413 - 422 из 718 на хромосоме X. Получено 1 сентября 2011 г.
- ^ MW King: Введение в болезнь Фабри. Статус: 13 февраля 2011 г., по состоянию на 1 сентября 2011 г.
- ↑ К. Фальке: Исследования микроструктуры вертициллярной роговицы при болезни Фабри и сравнение с лекарственно-индуцированной формой вертициллезной роговицы с помощью конфокальной микроскопии in vivo. Диссертация, Ростокский университет, 2009 г.
- ↑ Х. Ф. Уиллард: Половые хромосомы и инактивация Х-хромосомы. В: CR Scriver , AL Beaudet, WS Sly, D. Valle (Eds.): Метаболические и молекулярные основы наследственных заболеваний. Том 3, 8-е издание, MacGraw-Hill, 2001, ISBN 0-07-136322-X , стр. 1191-1211.
- ↑ а б в Л.Л. Пинто, Т.А. Виейра и др.: Проявление болезни на женщинах-носителях Х-сцепленных лизосомных нарушений: краткий обзор. В: Журнал редких заболеваний Orphanet. Том 5, 2010 г., стр. 14, DOI: 10.1186 / 1750-1172-5-14 . PMID 20509947 . PMC 2889886 (полный текст). (Обзор в открытом доступе )
- ^ A b Д.П. Жермен, К. Бенистан, Л. Ангелова: Х-сцепленное наследование и его значение в диагностике и ведении пациентов женского пола с болезнью Фабри. В: La Revue de médecine internal. Том 31 Дополнение 2, декабрь 2010 г., стр. S209-S213, DOI : 10.1016 / S0248-8663 (10) 70013-8 . PMID 21211665 .
- ↑ a b WB Dobyns, A. Filauro et al.: Наследование большинства X-сцепленных признаков не является доминантным или рецессивным, а только X-сцепленным. В: Американский журнал медицинской генетики. Часть A. Том 129A, номер 2, август 2004 г., стр. 136-143, DOI: 10.1002 / ajmg.a.30123 . PMID 15316978 .
- ^ WB Dobyns: Модель наследования признаков, сцепленных с Х-хромосомой, не является доминантной или рецессивной, а является лишь Х-сцепленной. В: Acta paediatrica. Том 95, номер 451, апрель 2006 г., стр. 11-15, DOI: 10.1111 / j.1651-2227.2006.tb02383.x . PMID 16720459 .
- ^ BR Migeon: инактивация X, женский мозаицизм и половые различия при почечных заболеваниях. В: ЯСН. Том 19, номер 11, ноябрь 2008 г., стр. 2052-2059, DOI: 10.1681 / ASN.2008020198 . PMID 18448583 . (Рассмотрение).
- ↑ М. Кобаяси, Т. Охаши и др.: Клинические проявления и естественное течение японских гетерозиготных женщин с болезнью Фабри. В: Журнал наследственных метаболических заболеваний. [электронная публикация перед печатью] январь 2008 г., DOI: 10.1007 / s10545-007-0740-6 . PMID 18202903 .
- ^ BR Migeon: Женщины - это мозаика: инактивация X и половые различия в болезнях. Нью-Йорк, Оксфордский университет, 2007, ISBN 978-0-19-518812-7 .
- ↑ I. Redonnet-Vernhet, JK Ploos van Amstel et al.: Неравномерная инактивация X в женской монозиготной паре близнецов с болезнью Фабри и дискордантная экспрессия новой мутации в гене альфа-галактозидазы А. В кн . : Журнал медицинской генетики. Том 33, номер 8, август 1996 г., стр. 682-688, PMID 8863162 . PMC 1050704 (полный текст).
- ↑ EM Maier, S. Osterrieder et al.: Проявления болезни и инактивация X у гетерозиготных женщин с болезнью Фабри. В: Acta paediatrica. Том 95, номер 451, апрель 2006 г., стр. 30–38, DOI: 10.1111 / j.1651-2227.2006.tb02386.x . PMID 16720462 .
- ↑ а б в А. Гал, Э. Шефер, И. Рохард: Генетические основы болезни Фабри. В: А. Мехта, М. Бек, Г. Сандер-Плассманн (ред.): Болезнь Фабри: перспективы 5 лет существования FOS. Глава 33, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290673
- ^ A. Gal: Молекулярная генетика болезни Фабри и корреляция генотип-фенотип. В: Д. Эльштейн, Г. Альтареску, М. Бек (ред.): Болезнь Фабри. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , стр. 3-19. ограниченный предварительный просмотр в поиске книг Google
- ^ RO Брэди : Болезнь Фабри - Обзор. В: Д. Эльштейн, Г. Альтареску, М. Бек (ред.): Болезнь Фабри. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , стр. XIX ограниченный предварительный просмотр в поиске книг Google
- ↑ a b MD Санчес-Ниньо, AB Sanz et al.: Действие глоботриаозилсфингозина на подоциты клубочков человека: значение для нефропатии Фабри. В кн . : Нефрология, диализ, трансплантация. Том 26, номер 6, июнь 2011 г., стр. 1797-1802, DOI: 10.1093 / ndt / gfq306 . PMID 20504837 .
- ↑ a b А. Х. Футерман, Г. ван Меер: Клеточная биология лизосомных нарушений накопления. В: Nature Reviews Molecular Cell Biology . Том 5, номер 7, июль 2004 г., стр. 554-565, DOI : 10.1038 / nrm1423 . PMID 15232573 . (Рассмотрение).
- ↑ М. Фуллер: Сфинголипиды: связь между болезнью Гоше и инсулинорезистентностью. В: Липиды в здоровье и болезни. Том 9, 2010 г., стр. 113, DOI: 10.1186 / 1476-511X-9-113 . PMID 20937139 . PMC 2964722 (полный текст). (Обзор в открытом доступе ).
- ↑ a b c AC Vedder, GE Linthorst et al.: Лечение болезни Фабри: результат сравнительного исследования с агалсидазой альфа или бета в дозе 0,2 мг / кг. В: PloS one. Том 2, номер 7, 2007 г., стр. E598, DOI : 10.1371 / journal.pone.0000598 . PMID 17622343 . PMC 1913555 (полный текст). ( Открытый доступ )
- ↑ KD MacDermot, A. Holmes, AH Miners: Естественная история болезни Фабри у пораженных мужчин и женщин-носителей. В: Журнал наследственных метаболических заболеваний. Volume 24 Suppl 2, 2001, pp. 13-14, PMID 11758673 .
- ↑ К. Уайбра, К. Кампманн и др.: Болезнь Андерсона-Фабри: клинические проявления болезни у гетерозигот женского пола. В: Журнал наследственных метаболических заболеваний. Том 24, номер 7, декабрь 2001 г., стр. 715-724, PMID 11804208 .
- ↑ PB Deegan, AF Baehner u a .: Естественная история болезни Фабри у женщин в обзоре результатов Фабри. В кн . : Журнал медицинской генетики. Том 43, номер 4, апрель 2006 г., стр. 347-352, DOI: 10.1136 / jmg.2005.036327 . PMID 16227523 . PMC 2563231 (полный текст).
- ↑ С. Гупта, М. Райс и др.: Связь накопления гликолипидов в сосудах с клиническими проявлениями болезни Фабри: поперечное исследование большой когорты гетерозиготных женщин с клиническим поражением. В кн . : Медицина. Том 84, номер 5, сентябрь 2005 г., стр. 261-268, PMID 16148726 .
- ↑ a b c AC Vedder, GE Linthorst et al.: Голландская когорта Фабри: разнообразие клинических проявлений и уровней Gb3. В: Журнал наследственных метаболических заболеваний. Том 30, номер 1, февраль 2007 г., стр. 68-78, DOI: 10.1007 / s10545-006-0484-8 . PMID 17206462 .
- ↑ AC Vedder, A. Strijland et al.: Проявления болезни Фабри в ткани плаценты. В: Журнал наследственных метаболических заболеваний. Том 29, номер 1, февраль 2006 г., стр. 106-111, DOI: 10.1007 / s10545-006-0196-0 . PMID 16601876 .
- ↑ а б Э. Янг, К. Миллс и др.: Является ли глоботриаозилцерамид полезным биомаркером болезни Фабри? В: Acta paediatrica. Volume 94, Number 447, March 2005, pp. 51-54, PMID 15895713 .
- ↑ С. Бекри, О. Лидов и др.: Роль церамида тригексозида (глоботриаозилцерамида) в диагностике и последующем контроле эффективности лечения болезни Фабри: обзор литературы. В кн . : Сердечно-сосудистые и гематологические агенты в медицинской химии. Том 4, номер 4, октябрь 2006 г., стр. 289-297, PMID 17073606 . (Рассмотрение).
- ↑ JM Aerts, JE Groener et al.: Повышенный уровень глоботриаозилсфингозина является признаком болезни Фабри. В: PNAS . Том 105, номер 8, февраль 2008 г., стр. 2812-2817, DOI : 10.1073 / pnas.0712309105 . PMID 18287059 . PMC 2268542 (полный текст).
- ↑ a b AH Miners, A. Holmes et al.: Оценка связанного со здоровьем качества жизни мужчин с болезнью Андерсона-Фабри до терапевтического вмешательства. В кн . : Исследование качества жизни. Volume 11, Number 2, March 2002, pp. 127-133, PMID 12018736 .
- ↑ a b Д.А. Лэйни, Д.Д. Грускин и др.: Социально-адаптивное и психологическое функционирование пациентов, страдающих болезнью Фабри. В: Журнал наследственных метаболических заболеваний. Январь 2010 г., DOI: 10.1007 / s10545-009-9025-6 . PMID 20087663 .
- ↑ a b AJ Grau, M. Schwaninger et al.: Лизосомное метаболическое заболевание с новыми терапевтическими возможностями. В кн . : Невролог. Том 74, номер 6, июнь 2003 г., стр. 489-496, DOI: 10.1007 / s00115-003-1513-6 . PMID 12799787 .
- ↑ Дж. Садек, Р. Шелл Хаас и др .: Психиатрические данные у четырех женщин-носителей болезни Фабри. В кн . : Психиатрическая генетика. Том 14, номер 4, декабрь 2004 г., стр. 199-201, PMID 15564893 .
- ↑ а б А.Л. Коул, П.Дж. Ли и др.: Депрессия у взрослых с болезнью Фабри: распространенная и недооцененная проблема В: Журнал наследственных метаболических заболеваний. Том 30, номер 6, ноябрь 2007 г., стр. 943-951, DOI : 10.1007 / s10545-007-0708-6 . PMID 17994284 .
- ↑ П. Сигал, Ю. Кон и др.: Психиатрический и когнитивный профиль у пациентов Андерсона-Фабри: предварительное исследование. В: Журнал наследственных метаболических заболеваний. Том 33, номер 4, август 2010 г., стр. 429-436, DOI : 10.1007 / s10545-010-9133-3 . PMID 20549363 .
- ↑ TW Crosbie, W. Packman, S. Packman: Психологические аспекты пациентов с болезнью Фабри. В: Журнал наследственных метаболических заболеваний. Том 32, номер 6, декабрь 2009 г., стр. 745-753, DOI : 10.1007 / s10545-009-1254-1 . PMID 19924564 .
- ↑ WJ Cable, EH Kolodny, RD Adams: Болезнь Фабри: нарушение вегетативной функции. В кн . : Неврология. Том 32, номер 5, май 1982 г., стр. 498-502, PMID 6803189 .
- ↑ М. Дютч, Х. Мартол и др.: Дисфункция мелких волокон преобладает при невропатии Фабри. В кн . : Журнал клинической нейрофизиологии. Том 19, номер 6, декабрь 2002 г., стр. 575-586, PMID 12488789 .
- ↑ Б. Хоффманн, М. Бек и др.: Природа и распространенность боли при болезни Фабри и ее ответ на заместительную ферментативную терапию - ретроспективный анализ исследования результатов Фабри. В: Клинический журнал боли. Том 23, номер 6, июль-август 2007 г., стр. 535-542, DOI: 10.1097 / AJP.0b013e318074c986 . PMID 17575495 .
- ↑ a b RJ Hopkin, J. Bissler et al.: Характеристика болезни Фабри у 352 педиатрических пациентов в реестре Фабри. В кн . : Педиатрические исследования. Том 64, номер 5, ноябрь 2008 г., стр. 550-555, DOI: 10.1203 / PDR.0b013e318183f132 . PMID 18596579 .
- ↑ Дж. Чарроу: 14-летний мальчик с болью в руках и ногах. В кн . : Педиатрические анналы. Том 38, номер 4, апрель 2009 г., стр. 190, 192, PMID 19455947 .
- ↑ MJ Hilz, B. Stemper, EH Kolodny: Воздействие холода на нижние конечности вызывает боль и длительную дисфункцию мелких волокон у пациентов с Fabry. В: Боль. Том 84, номер 2-3, февраль 2000 г., стр. 361-365, PMID 10666542 .
- ↑ KJ Sheth, SL Werlin et al.: Структура и функция желудочно-кишечного тракта при болезни Фабри. В: Американский журнал гастроэнтерологии. Том 76, номер 3, сентябрь 1981 г., стр. 246-251, PMID 6274188 .
- ↑ а б Б. Хоффманн, М. Шварц и др.: Желудочно-кишечные симптомы у 342 пациентов с болезнью Фабри: распространенность и ответ на заместительную ферментную терапию. В кн . : Клиническая гастроэнтерология и гепатология. Том 5, номер 12, декабрь 2007 г., стр. 1447-1453, DOI: 10.1016 / j.cgh.2007.08.012 . PMID 17919989 .
- ^ WH Kang, SI Chun, S. Lee: Общий ангидроз, связанный с болезнью Фабри. В: Журнал Американской академии дерматологии. Том 17, номер 5, часть 2, ноябрь 1987 г., стр. 883-887, PMID 3119682 .
- ↑ a b c d CH Ортеу, Т. Янсен и другие: Болезнь Фабри и кожа: данные FOS, итогового исследования Фабри. В: Британский дерматологический журнал. Том 157, номер 2, август 2007 г., стр. 331-337, DOI: 10.1111 / j.1365-2133.2007.08002.x . PMID 17573884 .
- ↑ а б С. Н. Гупта, М. Рис и др.: Импеданс кожи при болезни Фабри: проспективное контролируемое нерандомизированное клиническое исследование. В кн . : БМК неврология. Том 8, 2008 г., стр. 41, DOI: 10.1186 / 1471-2377-8-41 . PMID 18990229 . PMC 2585087 (полный текст). ( Открытый доступ )
- ^ A b ED Shelley, WB Shelley, TW Kurczynski: Болезненные пальцы, непереносимость тепла и телеангиэктазии уха: легко игнорировать детские признаки болезни Фабри. В кн . : Детская дерматология. Том 12, номер 3, сентябрь 1995 г., стр. 215-219, PMID 7501549 .
- ↑ CM Eng, DP Germain et al.: Болезнь Фабри: рекомендации по оценке и лечению поражения мультиорганной системы. В кн . : Генетика в медицине. Том 8, номер 9, сентябрь 2006 г., стр. 539-548, DOI : 10.1097 / 01.gim.0000237866.70357.c6 . PMID 16980809 . (Рассмотрение).
- ↑ М. Мёреншлагер, М. Браун-Фалько и др.: Болезнь Фабри: распознавание и лечение кожных проявлений. В: Американский журнал клинической дерматологии. Том 4, номер 3, 2003 г., стр. 189-196, PMID 12627994 . (Рассмотрение).
- ↑ Д. Ваттанасиричайгун, Дж. Свасти и др.: Клиническая и молекулярная характеристика расширенной семьи с болезнью Фабри. В: Журнал Медицинской ассоциации Таиланда. Выпуск 89, номер 9, сентябрь 2006 г., стр. 1528-1535, PMID 17100396 .
- ↑ К. Орссо, Ж. Дюфье, Д. Жермен: Глазные проявления при болезни Фабри: опрос 32 гемизиготных пациентов мужского пола. В кн . : Офтальмологическая генетика. Том 24, номер 3, сентябрь 2003 г., стр. 129-139, PMID 12868031 .
- ↑ Т. Т. Нгуен, Т. Гин и др.: Офтальмологические проявления болезни Фабри: опрос пациентов в Королевском центре лечения болезней Фабри в Мельбурне. В кн . : Клиническая и экспериментальная офтальмология. Том 33, номер 2, апрель 2005 г., стр. 164-168, DOI: 10.1111 / j.1442-9071.2005.00990.x . PMID 15807825 .
- ↑ А. Соди, А. С. Иоаннидис и др.: Глазные проявления болезни Фабри: данные Fabry Outcome Survey. В: Британский офтальмологический журнал. Том 91, номер 2, февраль 2007 г., стр. 210-214, DOI: 10.1136 / bjo.2006.100602 . PMID 16973664 . PMC 1857640 (полный текст).
- ↑ К. Фальке, А. Бюттнер и др.: Микроструктура вертициллезной роговицы при болезни Фабри и кератопатии, вызванной амиодароном: исследование конфокальной лазерной сканирующей микроскопии. В: Архив Грефе по клинической и экспериментальной офтальмологии. Том 247, номер 4, апрель 2009 г., стр. 523-534, DOI : 10.1007 / s00417-008-0962-9 . PMID 18931853 .
- ↑ А. Сесса, М. Мерони и др.: Патологические изменения почек при болезни Фабри. В: Журнал наследственных метаболических заболеваний. Volume 24 Suppl 2, 2001, pp. 66-70, PMID 11758681 . (Рассмотрение).
- ↑ А. Сесса, М. Мерони и др.: Эволюция почечной патологии при болезни Фабри. В: Acta paediatrica. Том 92, номер 443, декабрь 2003 г., стр. 6-8, PMID 14989458 .
- ↑ a b AB Fogo, L. Bostad et al.: Система оценки почечной патологии при болезни Фабри: отчет Международной исследовательской группы нефропатии Фабри (ISGFN). В кн . : Нефрология, диализ, трансплантация. Том 25, номер 7, июль 2010 г., стр. 2168-2177, DOI : 10.1093 / ndt / gfp528 . PMID 19833663 . PMC 2902894 (полный текст).
- ↑ MC Gubler, G. Lenoir et al.: Ранние почечные изменения у гемизиготных и гетерозиготных пациентов с болезнью Фабри. В: Kidney International . Том 13, номер 3, март 1978 г., стр. 223-235, PMID 418264 .
- ↑ Дж. Алрой, С. Сабнис, Дж. Б. Копп: Патология почек при болезни Фабри. В: ЯСН. Том 13 Дополнение 2, июнь 2002 г., стр. S134-S138, PMID 12068025 . (Рассмотрение).
- ↑ а б в г д Т. Томайдис, М. Релле и др.: Влияние заместительной ферментной терапии (ФЗТ) на функцию почек у пациентов с болезнью Фабри. В кн . : Медицинская клиника. Том 104, номер 9, сентябрь 2009 г., стр. 699-703, DOI : 10.1007 / s00063-009-1152-1 . PMID 19779674 . (Рассмотрение).
- ↑ А. Ортис, Дж. П. Оливейра и др.: Нефропатия у мужчин и женщин с болезнью Фабри: поперечное описание пациентов до лечения заместительной ферментной терапией. В кн . : Нефрология, диализ, трансплантация. Том 23, номер 5, май 2008 г., стр. 1600-1607, DOI: 10.1093 / ndt / gfm848 . PMID 18175781 .
- ↑ a b М. Х. Брантон, Р. Шиффманн и др.: Естественная история почечной болезни Фабри: влияние активности альфа-галактозидазы А и генетических мутаций на клиническое течение. В кн . : Медицина. Volume 81, Number 2, March 2002, pp. 122-138, PMID 11889412 .
- ↑ а б в г Р. Шиффманн, Д. Г. Варнок и др.: Болезнь Фабри: прогрессирование нефропатии и распространенность сердечных и цереброваскулярных событий до заместительной ферментативной терапии. В кн . : Нефрология, диализ, трансплантация. Том 24, номер 7, июль 2009 г., стр. 2102-2111, DOI : 10.1093 / ndt / gfp031 . PMID 19218538 . PMC 2698092 (полный текст).
- ↑ Glass RB et al. J Comput Assist Tomogr (2004) 28: 158-168.
- ↑ a b c А. Линхарт, Т. Палецек и др.: Новые сведения о структурных изменениях сердца у пациентов с болезнью Фабри. В: Американский журнал сердца . Том 139, номер 6, июнь 2000 г., стр. 1101-1108, DOI: 10.1067 / mhj.2000.105105 . PMID 10827394 .
- ↑ а б в К. Кампманн, Ф. Бэнер и др.: Кардиальные проявления болезни Андерсона-Фабри у гетерозиготных женщин. В: Журнал Американского колледжа кардиологии . Том 40, номер 9, ноябрь 2002 г., стр. 1668-1674, PMID 12427421 .
- ↑ а б М. Сенешаль, Д. П. Жермен: Болезнь Фабри: функциональное и анатомическое исследование сердечных проявлений у 20 гемизиготных пациентов мужского пола. В кн . : Клиническая генетика. Том 63, номер 1, январь 2003 г., стр. 46-52, PMID 12519371 .
- ^ A b JS Shah, DA Hughes et al.: Распространенность и клиническое значение сердечной аритмии при болезни Андерсона-Фабри. В кн . : Американский кардиологический журнал. Том 96, номер 6, сентябрь 2005 г., стр. 842-846, DOI : 10.1016 / j.amjcard.2005.05.033 . PMID 16169374 .
- ^ PM Elliott, H. Kindler et al.: Коронарная микрососудистая дисфункция у пациентов мужского пола с болезнью Андерсона-Фабри и эффект лечения альфа-галактозидазой A. В: Сердце. Том 92, номер 3, март 2006 г., стр. 357-360, DOI: 10.1136 / hrt.2004.054015 . PMID 16085718 . PMC 1860797 (полный текст).
- ↑ Дж. К. Мун, М. Шеппард и др.: Гистологическая основа сердечно-сосудистого магнитного резонанса с поздним увеличением гадолиния у пациента с болезнью Андерсона-Фабри. В: J Cardiovasc Magn Reson. Том 8, номер 3, 2006 г., стр. 479-482, PMID 16755835 .
- ↑ Х. Хасегава, Х. Такано и др.: Изображения в сердечно-сосудистой медицине. Переход от гипертрофии левого желудочка к массивному фиброзу при сердечном варианте болезни Фабри. В кн . : Тираж. Том 113, номер 16, апрель 2006 г., стр. E720 - e721, DOI : 10.1161 / CIRCULATIONAHA.105.584292 . PMID 16636179 .
- ↑ Ф. Вайдеманн, Ф. Бреуниг и др.: Вариации морфологических и функциональных сердечных проявлений при болезни Фабри: потенциальные последствия для временного течения болезни. В кн . : Европейский сердечный журнал. Том 26, номер 12, июнь 2005 г., стр. 1221-1227, DOI: 10.1093 / eurheartj / ehi143 . PMID 15728649 .
- ↑ А. Линхарт, К. Кампманн и др.: Кардиальные проявления болезни Андерсона-Фабри: результаты международного исследования результатов Фабри. В кн . : Европейский сердечный журнал. Том 28, номер 10, май 2007 г., стр. 1228-1235, DOI: 10.1093 / eurheartj / ehm153 . PMID 17483538 .
- ↑ К. Кампманн, А. Линхарт и др.: Начало и прогрессирование кардиомиопатии, связанной с болезнью Андерсона-Фабри. В кн . : Международный кардиологический журнал. Том 130, номер 3, ноябрь 2008 г., стр. 367-373, DOI : 10.1016 / j.ijcard.2008.03.007 . PMID 18572264 .
- ↑ a b T. Takenaka, H. Teraguchi и др.: Кардиологические находки на терминальной стадии у пациентов с сердечной болезнью Фабри: электрокардиографические, эхокардиографические и аутопсийные исследования. В кн . : Кардиологический журнал. Том 51, номер 1, февраль 2008 г., стр. 50-59, DOI: 10.1016 / j.jjcc.2007.12.001 . PMID 18522775 .
- ↑ a b M. Pieroni, C. Chimenti et al.: Раннее выявление кардиомиопатии Фабри с помощью тканевой доплеровской визуализации. В кн . : Тираж. Volume 107, Number 15, April 2003, pp. 1978-1984, DOI: 10.1161 / 01.CIR.0000061952.27445.A0 . PMID 12668521 .
- ^ F. Weidemann, F. Breunig и др.: Улучшение сердечной функции во время заместительной ферментной терапии у пациентов с болезнью Фабри: проспективное исследование с визуализацией скорости деформации. В кн . : Тираж. Выпуск 108, номер 11, сентябрь 2003 г., стр. 1299-1301, DOI: 10.1161 / 01.CIR.0000091253.71282.04 . PMID 12952834 .
- ↑ M. Pieroni, C. Chimenti et al .: Тканевая допплерография при болезни Фабри. В кн . : Современные взгляды в кардиологии. Том 19, номер 5, сентябрь 2004 г., стр. 452-457, PMID 15316452 . (Рассмотрение).
- ↑ Т. Палечек, Г. Досталова и др .: Поражение правого желудочка при болезни Фабри. В: Журнал Американского общества эхокардиографии. Том 21, номер 11, ноябрь 2008 г., стр. 1265-1268, DOI: 10.1016 / j.echo.2008.09.002 . PMID 18835697 .
- ↑ М. Ниманн, Ф. Бреуниг и др.: Правый желудочек при болезни Фабри: естественное течение и влияние заместительной ферментной терапии. В: Сердце. Том 96, номер 23, декабрь 2010 г., стр. 1915-1919, DOI: 10.1136 / hrt.2010.204586 . PMID 20965976 .
- ↑ К. Кампманн, Ф. А. Байнер и др.: Правый желудочек при болезни Фабри. В: Acta paediatrica. Том 94, номер 447, март 2005 г., стр. 15-18, PMID 15895706 .
- ↑ a b A. Fellgiebel, MJ Müller, L. Ginsberg: Проявления болезни Фабри со стороны ЦНС. В кн . : Ланцетная неврология. Том 5, номер 9, сентябрь 2006 г., стр. 791-795, DOI : 10.1016 / S1474-4422 (06) 70548-8 . PMID 16914407 .
- ↑ a b c К. Симс, Дж. Политей и др.: Инсульт при болезни Фабри часто возникает до постановки диагноза и при отсутствии других клинических проявлений: данные естественного анамнеза из реестра Фабри. В: Инсульт. Том 40, номер 3, март 2009 г., стр. 788-794, DOI : 10.1161 / STROKEAHA.108.526293 . PMID 19150871 .
- ↑ а б П. Мициас, С. Р. Левин: Цереброваскулярные осложнения болезни Фабри. В кн . : Анналы неврологии. Том 40, номер 1, июль 1996 г., стр. 8-17, DOI: 10.1002 / ana.410400105 . PMID 8687196 .
- ↑ MF Mendez, TM Stanley et al.: Сосудистая деменция при болезни Фабри. В: Деменция и гериатрические когнитивные расстройства. Том 8, номер 4, июль-август 1997 г., стр. 252-257, PMID 9213072 .
- ↑ Р. Океда, М. Нисихара: Случай вскрытия трупа болезни Фабри с невропатологическим исследованием патогенеза ассоциированной деменции. В кн . : Невропатология. Том 28, номер 5, октябрь 2008 г., стр. 532-540, DOI : 10.1111 / j.1440-1789.2008.00883.x . PMID 18410273 .
- ↑ А. Фелльгибель, И. Келлер и др.: Диагностическая ценность различных методов МРТ и МР-ангиографии при болезни Фабри. В кн . : Неврология. Том 72, номер 1, январь 2009 г., стр. 63-68, DOI: 10.1212 / 01.wnl.0000338566.54190.8a . PMID 19122032 .
- ↑ T. DeGraba, S. Azhar et al.: Профиль активации эндотелия и лейкоцитов у пациентов с Fabry. В кн . : Анналы неврологии. Том 47, номер 2, февраль 2000 г., стр. 229-233, PMID 10665494 .
- ↑ Д. Ф. Мур, Л. Т. Скотт и др.: Региональная церебральная гиперперфузия и нарушение регуляции пути оксида азота при болезни Фабри: отмена ферментной заместительной терапии. В кн . : Тираж . Том 104, номер 13, сентябрь 2001 г., стр. 1506-1512, PMID 11571244 .
- ↑ Д. Ф. Мур, Г. Альтареску и др.: Повышенная скорость церебрального кровотока при болезни Фабри с реверсией после замены ферментов. В: Инсульт; журнал мозгового кровообращения. Том 33, номер 2, февраль 2002 г., стр. 525-531, PMID 11823664 .
- ↑ Р. Шиффманн: Болезнь Фабри. В кн . : Фармакология и терапия . Том 122, номер 1, апрель 2009 г., стр. 65-77, DOI: 10.1016 / j.pharmthera.2009.01.003 . PMID 19318041 . (Рассмотрение).
- ↑ CR Kaneski, DF Moore et al.: Миелопероксидаза предсказывает риск васкулопатических событий у гемизгиготных мужчин с болезнью Фабри. В кн . : Неврология. Том 67, номер 11, декабрь 2006 г., стр. 2045-2047, DOI : 10.1212 / 01.wnl.0000247278.88077.09 . PMID 17159117 . PMC 1950664 (полный текст).
- ↑ А. Палла, С. Хегеманн и др.: Вестибулярный и слуховой дефицит при болезни Фабри и их ответ на заместительную ферментную терапию. В кн . : Журнал неврологии. Volume 254, Number 10, October 2007, pp. 1433-1442, DOI: 10.1007 / s00415-007-0575-y . PMID 17934877 .
- ^ Y. Сакураи, Х. Кодзима и др .: Статус слуха у 12 женщин и 15 мужчин японских пациентов Fabry. В: auris, nasus, гортань. Том 36, номер 6, декабрь 2009 г., стр. 627-632, DOI : 10.1016 / j.anl.2009.01.001 . PMID 19261412 .
- ↑ DP Germain, P. Avan et al.: Пациенты, страдающие болезнью Фабри, имеют повышенную частоту прогрессирующей потери слуха и внезапной глухоты: исследование двадцати двух гемизиготных пациентов мужского пола. В кн . : BMC Medical Genetics. Том 3, октябрь 2002 г., стр. 10, PMID 12377100 . PMC 134464 (полный текст).
- ^ Г. Конти, Б. Серджи: Слуховые и вестибулярные данные при болезни Фабри: исследование гемизиготных мужчин и гетерозиготных женщин. В: Acta paediatrica. Том 92, номер 443, декабрь 2003 г., стр. 33–37, PMID 14989464 .
- ↑ Д.М. Розенберг, В.Дж. Ферранс и др.: Хроническая обструкция воздушного потока при болезни Фабри. В: Американский журнал медицины. Выпуск 68, номер 6, июнь 1980 г., стр. 898-905, PMID 6247911 .
- ↑ Л.К. Браун, А. Миллер и др.: Поражение легких при болезни Фабри. В: Американский журнал респираторной медицины и реанимации. Volume 155, Number 3, March 1997, pp. 1004-1010, PMID 9116979 .
- ↑ S. Magage, JC Lubanda u a .: atteinte respiratoire de la maladie de Fabry. В кн . : Медицинские науки. Volume 21, Number 11 Suppl, декабрь 2005 г., стр. 37–39, DOI: 10.1051 / medsci / 20052111s37 . PMID 16324666 .
- ↑ S. Magage, JC Lubanda u a .: болезнь Естественная история респираторного поражения у Андерсона-Фабри. В: Журнал наследственных метаболических заболеваний. Том 30, номер 5, октябрь 2007 г., стр. 790-799, DOI : 10.1007 / s10545-007-0616-9 . PMID 17619837 .
- ↑ Д.П. Жермен, К. Бенистан и др.: Atteinte osseuse de la maladie de Fabry. В кн . : Медицинские науки. Volume 21, Number 11 Suppl, декабрь 2005 г., стр. 43-44, DOI: 10.1051 / medsci / 20052111s43 . PMID 16324668 .
- ↑ Д.П. Жермен, К. Бенистан и др.: Остеопения и остеопороз: ранее нераспознанные проявления болезни Фабри. В кн . : Клиническая генетика. Том 68, номер 1, июль 2005 г., стр. 93-95, DOI: 10.1111 / j.1399-0004.2005.00457.x . PMID 15952993 .
- ↑ Х. Мерсебах, Й.О. Йоханссон и др.: Остеопения: частый аспект болезни Фабри. Предикторы минеральной плотности костной ткани. В кн . : Генетика в медицине. Том 9, номер 12, декабрь 2007 г., стр. 812-818, DOI : 10.1097 / GIM.0b013e31815cb197 . PMID 18091430 .
- ^ RO Брэди : вовлечение костей и мышц в болезнь Фабри. В: Д. Эльштейн, Г. Альтареску, М. Бек (ред.): Болезнь Фабри. Verlag Springer, 2010, ISBN 978-90-481-9032-4 , стр. 293-298, ограниченный предварительный просмотр в поиске книг Google
- ↑ К.Л. Марчесони, Н. Роа и др.: Ошибочный диагноз при болезни Фабри. В кн . : Журнал педиатрии. Volume 156, Number 5, May 2010, pp. 828-831, doi: 10.1016 / j.jpeds.2010.02.012 . PMID 20385321 .
- ↑ А. Мехта, Р. Риччи и др.: Определение болезни Фабри: исходные клинические проявления у 366 пациентов в обзоре результатов Фабри. В: Европейский журнал клинических исследований. Том 34, номер 3, март 2004 г., стр. 236-242, DOI: 10.1111 / j.1365-2362.2004.01309.x . PMID 15025684 .
- ↑ а б в К. Д. МакДермот, А. Холмс, А. Х. Майнерс: Болезнь Андерсона-Фабри: клинические проявления и влияние болезни в когорте из 98 гемизиготных мужчин. В кн . : Журнал медицинской генетики. Том 38, номер 11, ноябрь 2001 г., стр. 750-760, PMID 11694547 . PMC 173476 (полный текст).
- ↑ Н. Зигмунд-Шульце: Болезнь Фабри встречается редко, симптомы неспецифичны - диагнозу часто предшествует одиссея от врача к врачу. В кн . : Газета «Врачи». от 15 декабря 2004 г.
- ↑ JS Mayes, JB Scheerer et al.: Дифференциальный анализ лизосомальных альфа-галактозидаз в тканях человека и его применение при болезни Фабри. В: Clinica chimica acta. Volume 112, Number 2, May 1981, pp. 247-251, PMID 6263521 .
- ↑ Б. Хоффманн, Х. Георг Кох и др.: Недостаточная активность альфа-галактозидазы А в плазме, но отсутствие болезни Фабри - ловушка в диагностике. В кн . : Клиническая химия и лабораторная медицина. Том 43, номер 11, 2005 г., стр. 1276-1277, DOI: 10.1515 / CCLM.2005.219 . PMID 16232095 .
- ↑ GE Linthorst, AC Vedder et al.: Скрининг на болезнь Фабри с использованием пятен цельной крови не позволяет выявить одну треть женщин-носителей. В: Clinica chimica acta. Том 353, номер 1-2, март 2005 г., стр. 201-203, DOI : 10.1016 / j.cccn.2004.10.019 . PMID 15698608 .
- ↑ а б Б.Е. Смид, Л. ван дер Тол, М. Бигстраатен, Г. Э. Линторст, ЦЕМ Холлак, Б. Дж. Х. М. Пуртуис. В: J Med Genet , 2015, 52 (4), стр. 262-226.
- ^ С. Рой, К. Годин и др.: Оптимизация разделения четырех основных нейтральных гликосфинголипидов: приложение для быстрого и простого обнаружения глоботриаозилцерамида в моче при болезни Фабри. В: Журнал хроматографии B. Том 805, номер 2, июнь 2004 г., стр. 331-337, DOI: 10.1016 / j.jchromb.2004.03.037 . PMID 15135109 .
- ↑ C. Auray-Blais, D. Cyr et al.: Разработка метода фильтровальной бумаги, потенциально применимого для массового скрининга мочи с высоким риском на болезнь Фабри. В: Журнал наследственных метаболических заболеваний. Том 30, номер 1, февраль 2007 г., стр. 106, DOI: 10.1007 / s10545-006-0444-3 . PMID 17171433 .
- ↑ C. Auray-Blais, D. Cyr et al.: Экскреция глоботриаозилцерамида с мочой коррелирует с генотипом у детей и взрослых с болезнью Фабри. В кн . : Молекулярная генетика и метаболизм. Том 93, номер 3, март 2008 г., стр. 331-340, DOI : 10.1016 / j.ymgme.2007.10.001 . PMID 18023222 .
- ↑ Д. Тубул, С. Рой и др.: Быстрое снятие отпечатков пальцев с помощью масс-спектрометрии MALDI-TOF гликосфинголипидов осадка мочи при болезни Фабри. В кн . : Аналитическая и биоаналитическая химия. Том 382, номер 5, июль 2005 г., стр. 1209-1216, DOI: 10.1007 / s00216-005-3239-8 . PMID 15959771 .
- ↑ К. Миллс, П. Моррис и др.: Измерение CDH и CTH в моче с помощью тандемной масс-спектрометрии у гемизиготных и гетерозиготных по болезни Фабри пациентов. В: Журнал наследственных метаболических заболеваний. Том 28, номер 1, 2005 г., стр. 35-48, DOI: 10.1007 / s10545-005-5263-4 . PMID 15702404 .
- ↑ Р.Дж. Десник: Пренатальная диагностика болезни Фабри. В кн . : Пренатальная диагностика. Том 27, номер 8, август 2007 г., стр. 693-694, DOI: 10.1002 / pd.1767 . PMID 17533632 . (Рассмотрение).
- ↑ К. Линденгрюн: Редкие недуги - Лица мукополисахаридозов. В: CliniCum. 9, 2010.
- ↑ AM Das: Врожденные нарушения обмена веществ. В: А. Ридер, Б. Лофф (ред.): Гендерная медицина. Verlag Springer, 2004, ISBN 3-211-00766-0 , стр. 67f. ограниченный предварительный просмотр в поиске книг Google
- ↑ TF Metz, TP Mechtler u a .: Упрощенный протокол скрининга новорожденных на лизосомные нарушения накопления. В кн . : Клиническая химия. Том 57, номер 9, сентябрь 2011 г., стр. 1286-1294, DOI: 10.1373 / Clinchem.2011.164640 . PMID 21771947 .
- ↑ К. Накамура, К. Хаттори, Ф. Эндо: Скрининг новорожденных на лизосомные нарушения накопления. В: Американский журнал медицинской генетики. Том 157, номер 1, февраль 2011 г., стр. 63-71, DOI : 10.1002 / ajmg.c.30291 . PMID 21312327 . (Рассмотрение).
- ↑ Спада М., Каспер Д., Пальярдини В., Биамино Э, Джахеро С., Порта Ф. Итал. J Pediatr 2017; 43 (1): 1
- ^ GM Pastores: Miglustat: субстратная терапия для расстройств лизосомного накопления, связанных с первичным поражением центральной нервной системы. ( Памятка от 16 июня 2010 г. в Интернет-архиве ) (PDF; 75 kB) В: Последние патенты на открытие лекарств для ЦНС. 1, 2006, с. 77-82. PMID 18221193
- ↑ а б Терапия болезни Фабри. ( Памятка от 3 февраля 2015 г. в Интернет-архиве ) Проверено 13 сентября 2011 г.
- ↑ Дж. М. Доннелли: Шайр получает одобрение FDA на лекарство от редких заболеваний. В: Бостонский деловой журнал. от 25 августа 2011 г.
- ↑ a b К.А. Блэк: Идите вперед, а не внутрь. В: Природная медицина. Том 17, номер 5, май 2011 г., стр. 515, DOI: 10.1038 / nm0511-515 . PMID 21546944 .
- ↑ a b М. Бек: Агалсидаза альфа - препарат для заместительной ферментной терапии при болезни Андерсона-Фабри. В кн . : Мнение экспертов по исследуемым препаратам. Том 11, номер 6, июнь 2002 г., стр. 851-858, DOI: 10.1517 / 13543784.11.6.851 . PMID 12036428 . (Рассмотрение).
- ↑ K. Lee, X. Jin et al.: Биохимическое и фармакологическое сравнение ферментативной заместительной терапии при болезни Фабри, связанной с нарушением накопления гликолипидов. В кн . : Гликобиология. Том 13, номер 4, апрель 2003 г., стр. 305-313, DOI: 10.1093 / glycob / cwg034 . PMID 12626384 .
- ↑ Специализированная информация Fabrazyme 5 мг, 35 мг, по состоянию на 11/2018
- ↑ a b WR Wilcox, M. Banikazemi et al.: Долгосрочная безопасность и эффективность заместительной ферментной терапии при болезни Фабри. В: Американский журнал генетики человека. Том 75, номер 1, июль 2004 г., стр. 65-74, DOI: 10.1086 / 422366 . PMID 15154115 . PMC 1182009 (полный текст).
- ↑ Б.Л. Турберг, Х. Реннке и др.: Накопление глоботриаозилцерамида в почках Фабри выводится из нескольких типов клеток после ферментативной заместительной терапии. В: Kidney International . Том 62, номер 6, декабрь 2002 г., стр. 1933-1946, DOI: 10.1046 / j.1523-1755.2002.00675.x . PMID 12427118 .
- ↑ a b c d Дж. Маршалл, К. М. Эш и др.: Уменьшение количества субстратов увеличивает эффективность ферментной терапии на мышиной модели болезни Фабри. В: PloS one. Том 5, номер 11, 2010 г., стр. E15033, DOI : 10.1371 / journal.pone.0015033 . PMID 21124789 . PMC 2991350 (полный текст). ( Открытый доступ )
- ↑ М. Бек: Агалсидаза альфа для лечения болезни Фабри: новые данные о клинической эффективности и безопасности. В кн . : Мнение экспертов по биологической терапии. Том 9, номер 2, февраль 2009 г., стр. 255-261, DOI: 10.1517 / 14712590802658428 . PMID 19236256 . (Рассмотрение).
- ↑ a b CM Eng, N. Guffon et al.: Безопасность и эффективность рекомбинантной человеческой альфа-галактозидазы A - заместительная терапия при болезни Фабри. В: Медицинский журнал Новой Англии . Том 345, номер 1, июль 2001 г., стр. 9–16, DOI: 10.1056 / NEJM200107053450102 . PMID 11439963 .
- ↑ GM Китинг, Д. Симпсон: Взгляд на бета-агалсидазу при болезни Фабри. В: BioDrugs. Volume 21, Number 4, 2007, pp. 269-271, PMID 17628124 . (Рассмотрение).
- ↑ DP Germain, S. Waldek et al.: Устойчивая долгосрочная стабилизация почек после 54 месяцев терапии агалсидазой бета у пациентов с болезнью Фабри. В: ЯСН. Том 18, номер 5, май 2007 г., стр. 1547-1557, DOI: 10.1681 / ASN.2006080816 . PMID 17409312 .
- ↑ а б Я. Таджима, И. Кавашима и др.: Использование модифицированной альфа-N-ацетилгалактозаминидазы в разработке заместительной ферментной терапии болезни Фабри. В: Американский журнал генетики человека. Том 85, номер 5, ноябрь 2009 г., стр. 569-580, DOI : 10.1016 / j.ajhg.2009.09.016 . PMID 19853240 . PMC 2775840 (полный текст).
- ↑ Т. Охаши, М. Сакума и др.: Влияние образования антител на снижение уровня глоботриаозилцерамида (GL-3) в моче пациентов Fabry во время терапии агалсидазой бета. В кн . : Молекулярная генетика и метаболизм. Том 92, номер 3, ноябрь 2007 г., стр. 271-273, DOI : 10.1016 / j.ymgme.2007.06.013 . PMID 17689998 .
- ↑ Т. Охаши, С. Иизука и др.: Снижение активности фермента альфа-Gal A в клетках фибробластов Фабри и тканях мышей Фабри, индуцированное сывороткой от антител-положительных пациентов с болезнью Фабри. В кн . : Молекулярная генетика и метаболизм. Том 94, номер 3, июль 2008 г., стр. 313-318, DOI : 10.1016 / j.ymgme.2008.03.008 . PMID 18456533 .
- ↑ Lenders et al., J Allergy and Clin. Иммунология, 2018
- ↑ Маартен Арендс, Мариеке Бигстраатен и др.: Агалсидаза альфа по сравнению с агалсидазой бета для лечения болезни Фабри: международное когортное исследование. В: Журнал медицинской генетики , 55, 2018 г., стр. 351, DOI: 10.1136 / jmedgenet-2017-104863 .
- ↑ Фармацевтическая газета, выпуск 05, 2016 г.
- ↑ a b c С.Д. Джеймс: Пациенты с болезнью Фабри заболевают, когда наркотики уходят за границу. В: abcNews.com. от 30 августа 2011 г.
- ↑ CA Black: Вступать или не входить. В: Природная медицина. Том 17, номер 8, 2011 г., стр. 921, DOI: 10.1038 / nm.2440 . PMID 21818082 .
- ↑ rme: EMEA: Нормирование церезима и фабразима. ( Страница больше не доступна , поиск в веб-архивах ) Информация: ссылка была автоматически помечена как дефектная. Пожалуйста, проверьте ссылку в соответствии с инструкциями, а затем удалите это уведомление. В: Aerzteblatt.de. от 28 июня 2009 г.
- ↑ Нехватка поставок церезима и фабразима - рекомендуется приоритетный доступ для пациентов, наиболее нуждающихся в лечении. (PDF; 35 kB) Пресс-релиз EMEA от 25 июня 2009 г.
- ↑ Г. Шу, К. Нуссбаум и др.: Работа с ноу-хау в международном сотрудничестве в области НИОКР. (PDF; 1,8 МБ) В: Федеральное министерство образования и науки (ред.), 2009, с. 21.
- ^ A b Е. Долгин: NIH стоит перед приказом по поводу нехватки лекарств для сирот. В кн . : Природная медицина. Том 17, номер 5, май 2011 г., стр. 522, DOI: 10.1038 / nm0511-522b . PMID 21546953 .
- ↑ М. Ратнер: Марш наркотиков Фабри отрицан. В кн . : Природная биотехнология . Том 29, номер 97, 2011 г., стр. 676, DOI: 10.1038 / nbt0211-97b .
- ^ Ф. С. Коллинз: Определение в случае фабразима, произведенного Genzyme Corporation. (PDF; 544 kB) С 1 декабря 2010 г.
- ^ Т. Стэйтон: Genzyme приносит свои извинения за последнюю задержку с Fabrazyme. В: FiercePharma. от 8 сентября 2011 г.
- ↑ T. Staton: Проблема контроля качества нарушает поток Fabrazyme. В: FiercePharma. от 5 августа 2011 г.
- ↑ a b Genzyme санофи начинает поставки препарата против болезни Фабри. В: Pharma Times. 2 марта 2012, доступ к 30 апреля 2019 .
- ↑ a b Shire отзывает одобрение FDA. Врачи газета , 16 марта 2012, доступ к 29 апреля 2019 .
- ↑ Shire отзывает Replagal в США, поскольку FDA хочет больше испытаний. Pharma Таймс, 15 марта 2012, доступ к 29 апреля 2019 .
- ↑ Хронология выпуска Fabrazyme, Replagal *. (PDF) 14 июля 2014, доступ к 29 апреля 2019 .
- ↑ U. Fricke, U. Schwabe: Neue Arzneimittel 2007. In: U. Schwabe, D. Paffrath (Ed.): Drug Ordinance Report 2008. Verlag Springer, 2008, стр. 74. Ограниченный предварительный просмотр в поиске книг Google
- ↑ Б. Хоффманн, Э. Маятепек: Болезнь Фабри - часто наблюдается, редко диагностируется. В: Deutsches Ärzteblatt international. Том 106, номер 26, июнь 2009 г., стр. 440–447, DOI: 10.3238 / arztebl.2009.0440 . PMID 19623315 . PMC 270439 (полный текст). (Рассмотрение).
- ↑ Т. Ватт, А.П. Бурлина и др.: Лечение агалсидазой бета связано с улучшением качества жизни пациентов с болезнью Фабри: данные реестра Фабри. В кн . : Генетика в медицине. Том 12, номер 11, ноябрь 2010 г., стр. 703-712, DOI : 10.1097 / GIM.0b013e3181f13a4a . PMID 20885332 .
- ↑ MR Filling-Katz, HF Merrick et al.: Карбамазепин при болезни Фабри: эффективное обезболивание с дозозависимым обострением вегетативной дисфункции. В кн . : Неврология . Volume 39, Number 4, April 1989, pp. 598-600, PMID 2494569 .
- ↑ Национальный институт здоровья и клинического совершенства: Невропатическая боль: фармакологическое лечение нейропатической боли у взрослых в неспециализированных условиях (клиническое руководство NICE 96). Центр клинической практики NICE; 2010 г.
- ^ Р. Барон, А. Биндер, Г. Васнер: невропатическая боль: диагностика, патофизиологические механизмы и лечение. В кн . : Ланцетная неврология. Том 9, номер 8, август 2010 г., стр. 807-819, DOI : 10.1016 / S1474-4422 (10) 70143-5 . PMID 20650402 . (Рассмотрение).
- ↑ G. Cruccu, C. Sommer et al.: Руководство EFNS по оценке нейропатической боли: пересмотрено в 2009 г. В: Европейский журнал неврологии. Том 17, номер 8, август 2010 г., стр. 1010-1018, DOI: 10.1111 / j.1468-1331.2010.02969.x . PMID 20298428 . (Рассмотрение).
- ↑ CE Argoff, NW Barton et al.: Желудочно-кишечные симптомы и задержка опорожнения желудка при болезни Фабри: ответ на метоклопрамид. В кн . : Связь ядерной медицины. Том 19, номер 9, сентябрь 1998 г., стр. 887-891, PMID 10581595 .
- ↑ Х. Тахир, Л.Л. Джексон, Д.Г. Варнок: Антипротеинурическая терапия и фабри-нефропатия: устойчивое снижение протеинурии у пациентов, получающих заместительную ферментную терапию агалсидазой-бета. В: ЯСН. Том 18, номер 9, сентябрь 2007 г., стр. 2609-2617, DOI: 10.1681 / ASN.2006121400 . PMID 17656478 .
- ↑ a b CE Stetter: Исследование сердечного метаболизма при болезни Фабри in vivo с использованием 31 фосфорной МР-спектроскопии. Диссертация, Университет Юлиуса Максимилиана, Вюрцбург, 2007.
- ↑ IB Tomasic, MC Metcalf et al.: Взаимное преобразование специфичностей лизосомальных ферментов человека, связанных с болезнями Фабри и Шиндлера. В кн . : Журнал биологической химии . Том 285, номер 28, июль 2010 г., стр. 21560-21566, DOI : 10.1074 / jbc.M110.118588 . PMID 20444686 . PMC 2898384 (полный текст).
- ↑ Галафольд. В: EMA Europe. 17 сентября 2018, доступ к 2 апреля 2019 .
- ↑ S. Ishii, HH Chang et al.: Мутантные ферменты альфа-галактозидазы A, идентифицированные у пациентов с болезнью Фабри с остаточной ферментативной активностью: биохимическая характеристика и восстановление нормального внутриклеточного процессинга с помощью 1-дезоксигалактоноджиримицина. В кн . : Биохимический журнал. Том 406, номер 2, сентябрь 2007 г., стр. 285-295, DOI : 10.1042 / BJ20070479 . PMID 17555407 . PMC 1948963 (полный текст).
- ↑ S. Ishii, HH Chang et al.: Доклиническая эффективность и безопасность 1-дезоксигалактоноджиримицина у мышей при болезни Фабри. В кн . : Журнал фармакологии и экспериментальной терапии. Том 328, номер 3, март 2009 г., стр. 723-731, DOI : 10.1124 / jpet.108.149054 . PMID 19106170 .
- ↑ Р. Хаманака, Т. Шинохара и др.: Спасение мутантной альфа-галактозидазы А в эндоплазматическом ретикулуме с помощью 1-дезоксигалактоноджиримицина приводит к перемещению в лизосомы. В: Biochimica et biophysica acta . Том 1782, номер 6, июнь 2008 г., стр. 408-413, DOI : 10.1016 / j.bbadis.2008.03.001 . PMID 18381081 .
- ↑ а б С. Бьастофф, Б. Дрегер: Calystegines. В: Г. А. Корделл (ред.): Алкалоиды: химия и биология. С. 91. Ограниченный предварительный просмотр в поиске Google Книг.
- ↑ Г. Х. Ям, Н. Босхард и др.: Фармакологический шаперон корректирует лизосомное накопление при болезни Фабри, вызванной некомпетентными по трафику вариантами. В кн . : Американский физиологический журнал. Том 290, номер 4, апрель 2006 г., стр. C1076-C1082, DOI : 10.1152 / ajpcell.00426.2005 . PMID 16531566 .
- ↑ Н. Асано, С. Исии и др.: Ингибирование и внутриклеточное усиление лизосомальной альфа-галактозидазы А в лимфобластах Фабри с помощью 1-дезоксигалактоноджиримицина и его производных in vitro. В: Европейский журнал биохимии . Volume 267, Number 13, July 2000, pp. 4179-4186, PMID 10866822 .
- ^ A b Г. Андреотти, MR Guarracino и др.: Прогнозирование реакции на фармакологические шапероны: лизосомальная альфа-галактозидаза человека, пример исследования. В: Журнал редких заболеваний Orphanet. Том 5, 2010 г., стр. 36, DOI: 10.1186 / 1750-1172-5-36 . PMID 21138548 . PMC 3016270 (полный текст). ( Открытый доступ )
- ↑ Р. Ханна, Р. Соска и др.: Фармакологический шаперон 1-дезоксигалактоноджиримицин снижает уровни тканевого глоботриаозилцерамида на мышиной модели болезни Фабри. В кн . : Молекулярная терапия. Том 18, номер 1, январь 2010 г., стр. 23-33, DOI: 10.1038 / mt.2009.220 . PMID 19773742 . PMC 2839206 (полный текст).
- ↑ Клиническое исследование (фаза III): Исследование эффектов перорального AT1001 (гидрохлорида мигаластата) у пациентов с болезнью Фабри на сайте Clinicaltrials.gov Национального института здоровья.
- ↑ Решение Комиссии от 22-V-2006 о признании препарата «1-дезоксигалактоноджиримицина гидрохлорид» орфанным препаратом в соответствии с Регламентом (ЕС) № 141/2000 Европейского парламента и Совета. (PDF; 17 kB) (PDF) 22 мая 2006 г.
- ↑ SH Shin, S. Kluepfel steel u. A .: Прогнозирование ответа мутированной альфа-галактозидазы A на фармакологический шаперон. В кн . : Фармакогенетика и геномика. Том 18, номер 9, сентябрь 2008 г., стр. 773-780, DOI : 10.1097 / FPC.0b013e32830500f4 . PMID 18698230 . PMC 2657085 (полный текст).
- ↑ А. Абэ, С. Грегори и др.: Снижение уровня глоботриаозилцерамида у мышей с болезнью Фабри путем лишения субстрата. В: Журнал клинических исследований. Том 105, номер 11, июнь 2000 г., стр. 1563-1571, DOI: 10.1172 / JCI9711 . PMID 10841515 . PMC 300859 (полный текст).
- ↑ a b c T. Heare, NJ Alp et al.: Тяжелая эндотелиальная дисфункция аорты на мышиной модели болезни Фабри; частичная профилактика лечением N-бутилдезоксиноджиримицином. В: Журнал наследственных метаболических заболеваний. Том 30, номер 1, февраль 2007 г., стр. 79-87, DOI : 10.1007 / s10545-006-0473-y . PMID 17189993 .
- ↑ EMA Europe: Zavesca. 17 сентября 2018, доступ к 2 апреля 2019 .
- ↑ GM Pastores, NL Barnett, EH Kolodny: открытое несравнительное исследование миглустата при болезни Гоше I типа: эффективность и переносимость в течение 24 месяцев лечения. В кн . : Клиническая терапия. Том 27, номер 8, август 2005 г., стр. 1215-1227, DOI: 10.1016 / j.clinthera.2005.08.004 . PMID 16199246 .
- ↑ CE Hollak, D. Hughes et al.: Miglustat (Zavesca) при болезни Гоше 1 типа: 5-летние результаты пост-авторизационной программы наблюдения за безопасностью. В кн . : Фармакоэпидемиология и безопасность лекарственных средств. Том 18, номер 9, сентябрь 2009 г., стр. 770-777, DOI : 10.1002 / pds.1779 . PMID 19507165 .
- ↑ а б Т. Охима, Р. Шиффманн и др.: Старение усиливается, а трансплантация костного мозга улучшает метаболические нарушения у мышей с болезнью Фабри. В: PNAS. Volume 96, Number 11, May 1999, pp. 6423-6427, PMID 10339603 . PMC 26897 (полный текст).
- ↑ T. Takenaka, GJ Murray et al.: Долгосрочная ферментативная коррекция и снижение липидов во многих органах мышей Fabry с первичной и вторичной трансплантацией, получающих трансдуцированные клетки костного мозга. В: PNAS. Выпуск 97, номер 13, июнь 2000 г., стр. 7515-7520, DOI : 10.1073 / pnas.120177997 . PMID 10840053 . PMC 16577 (полный текст).
- ↑ T. Yokoi, H. Kobayashi et al.: Минимальная потребность в донорских клетках для уменьшения хранения гликолипидов после трансплантации костного мозга на мышиной модели болезни Фабри. В кн . : Журнал генной медицины. Том 13, номер 5, май 2011 г., стр. 262-268, DOI : 10.1002 / jgm.1566 . PMID 21520359 .
- ↑ Ким С.Х., Роббинс П.Д .: Ретровирусные векторы для генной терапии. В: Дж. А. Баррангер, М. А. Кабрера-Салазар (ред.): Лизосомные нарушения накопления. Verlag Springer, 2007, ISBN 978-0-387-70908-6 , стр. 53-63. ограниченный предварительный просмотр в поиске книг Google
- ^ Дж. Парк, Дж. Дж. Мюррей и др.: Долгосрочная коррекция хранения глоботриаозилцерамида у мышей Fabry путем переноса генов, опосредованного рекомбинантным аденоассоциированным вирусом. В: PNAS. Volume 100, Number 6, March 2003, pp. 3450-3454, DOI : 10.1073 / pnas.0537900100 . PMID 12624185 . PMC 152313 (полный текст).
- ↑ Г. Цинь, Т. Такенака и др.: Преселективная генная терапия болезни Фабри. В: PNAS. Том 98, номер 6, март 2001 г., стр. 3428-3433, DOI : 10.1073 / pnas.061020598 . PMID 11248095 . PMC 30670 (полный текст).
- ^ JE Scherberich: Наследственные болезни почек. В: Г. Стейнбек, Г. Паумгартнер: Терапия внутренних болезней. Verlag Springer, 2005, ISBN 3-540-23750-X , стр. 576. Ограниченный предварительный просмотр в поиске книг Google
- ↑ YA Ioannou, KM Zeidner et al.: Болезнь Фабри: доклинические исследования демонстрируют эффективность замены альфа-галактозидазы A у мышей с дефицитом фермента. В: Американский журнал генетики человека. Том 68, номер 1, январь 2001 г., стр. 14-25, DOI: 10.1086 / 316953 . PMID 11115376 . PMC 1234907 (полный текст).
- ^ ME Haskins, U. Giger, DF Patterson: Животные модели лизосомных болезней накопления: их развитие и клиническая значимость. В: А. Мехта, М. Бек, Г. Сандер-Плассманн (ред.): Болезнь Фабри: перспективы 5 лет FOS. Глава 6, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X , PMID 21290677
- ↑ а б К.Д. МакДермот, А. Холмс, А. Х. Майнерс: Болезнь Андерсона-Фабри: клинические проявления и влияние болезни на когорту из 60 женщин-облигатных носителей. В кн . : Журнал медицинской генетики. Том 38, номер 11, ноябрь 2001 г., стр. 769-775, PMID 11732485 . PMC 1734754 (полный текст).
- ↑ А. Мехта, Дж. Т. Кларк и др.: Естественное течение болезни Фабри: изменение модели причин смерти в FOS - Fabry Outcome Survey. В кн . : Журнал медицинской генетики. Том 46, номер 8, август 2009 г., стр. 548-552, DOI: 10.1136 / jmg.2008.065904 . PMID 19473999 .
- ↑ а б в г д Х. Фабри: Исторический обзор болезни Фабри. В: Журнал наследственных метаболических заболеваний. Volume 24 Suppl 2, 2001, pp. 3-7, PMID 11758677 .
- ↑ а б в Дж. Фабри: Вклад в изучение узловатой геморрагической пурпуры (Purpura papulosa haemorrhagica Hebrae). В: Архив дерматологии и сифилиса . Том 42, 1898 г., стр. 187-200. DOI : 10.1007 / BF01986897
- ↑ а б в Т. Янсен, Ф. Г. Бехара, П. Альтмейер: Ангиокератом - клиника, диагностика и терапия. TKT - Европа 5S, 2003 г., ISBN 3-00-011357-6 .
- ↑ а б Дж. Фабри: О клинике и этиологии ангиокератомы. В: Архив дерматологии и сифилиса. Том 123, номер 2, 1916, стр. 294-307.
- ↑ Дж. Фабри: Еще один вклад в клинику наивиформной ангиокератомы (Naevus angiokeratosus). В: Dermacol Wochenschr. 90, 1930, стр. 339-341.
- ↑ В. Андерсон: Случай «Анджио-кератомы». В: Br J Dermatol. Том 10, 1898 г., стр. 113-117. DOI: 10.1111 / j.1365-2133.1898.tb16317.x
- ^ HJ Wallace: Angiokeratoma corporis diffusum. В: Br J Dermatol. 70, 1968, стр. 354-360. DOI : 10.1111 / j.1365-2133.1958.tb13796.x PMID 13584689
- ↑ W. Cottle: Бородавчатые наросты. В: Отчеты больницы Святого Георгия. 9, 1877–1878, стр. 753–762.
- ↑ J. Weicksel: Angiomatosis или Angiokeratosis universalis (очень редкое заболевание кожи и сосудов). В: Dtsch Med Wochenschr. Том 51, 1925, стр. 898-900.
- ↑ AW Pompen, M. Ruiter, HJ Wyers: Angiokeratoma corporis diffusum (universal) Fabry, как признак неизвестного внутреннего заболевания; два отчета о вскрытии. В: Acta medica Scandinavica. Том 128, номер 3, июнь 1947 г., стр. 234-255, PMID 18897399 .
- ↑ П. Рейтер: Большой медицинский словарь Springer. Verlag Springer, 2004, ISBN 3-540-21352-X , стр. 1186. Ограниченный предварительный просмотр в поиске книг Google
- ↑ К. Скриба: О патогенезе Angiokeratoma corporis diffusum Fabry с комплексом кардиовазоренальных симптомов. В: Verh Dtsch Ges Pathol. 34, 1950, стр. 221-226.
- ↑ Х. Хорнбостел, К. Скриба: Для диагностики ангиокератомы Фабри с комплексом сердечно-сосудистых симптомов как болезни накопления фосфатидов путем иссечения кожи. В: Клинический еженедельник. Том 31, номера 3-4, январь 1953 г., стр. 68-69, PMID 13062573 .
- ^ JR Колли, DL Миллер и др.: Поражение почек при диффузной ангиокератоме. В: Британский медицинский журнал. Том 1, номер 5082, май 1958 г., стр. 1266-1268, PMID 13536474 . PMC 2028866 (полный текст).
- ↑ CC Sweeley, B. Klionsky: Болезнь Фабри. Классификация как сфинголипидоз и частичная характеристика нового гликолипида. В кн . : Журнал биологической химии. Том 238, сентябрь 1963 г., стр. 3148-3150, PMID 14081947 .
- ↑ JM Opitz, FC Stiles et al.: Генетика Angiokeratoma Corporis Diffusum (болезнь Фабри) и ее взаимосвязь с локусом Xg. В: Американский журнал генетики человека. Том 17, номер 4, июль 1965 г., стр. 325-342, PMID 17948499 . PMC 1932618 (свободный полный текст).
- ↑ К. Хашимото, Б. Г. Гросс, В. Ф. Рычаг: Angiokeratoma Corporis Diffusum (Fabry). Гистохимические и электронно-микроскопические исследования кожи. В кн . : Журнал исследовательской дерматологии. Том 44, февраль 1965 г., стр. 119-128, DOI : 10.1038 / jid.1965.22 PMID 14258908 .
- ↑ М. Ниманн: Влияние длительной заместительной ферментной терапии на морфологию и функцию левого желудочка у пациентов с болезнью Фабри. Диссертация, Вюрцбургский университет Юлиуса Максимилиана, 2009.
- ^ RO Brady , AE Gal et al .: Ферментативный дефект при болезни Фабри. Дефицит церамид тригексозидазы. В: Медицинский журнал Новой Англии . Volume 276, Number 21, May 1967, pp. 1163-1167, DOI: 10.1056 / NEJM196705252762101 . PMID 6023233 .
- ↑ JA Kint: Болезнь Фабри: дефицит альфа-галактозидазы. В кн . : Наука . Volume 167, Number 922, February 1970, pp. 1268-1269, PMID 5411915 .
- ↑ JW Kusiak, JM Quirk, RO Brady : Очистка и свойства двух основных изоферментов альфа-галактозидазы из плаценты человека. В кн . : Журнал биологической химии . Том 253, номер 1, январь 1978 г., стр. 184-190, PMID 201618 .
- ↑ RJ Desnick, KY Allen et al.: Болезнь Фабри: ферментативная диагностика гемизигот и гетерозигот. Активность альфа-галактозидазы в плазме, сыворотке, моче и лейкоцитах. В кн . : Журнал лабораторной и клинической медицины. Том 81, номер 2, февраль 1973 г., стр. 157-171, PMID 4683418 .
- ^ RO Brady , JF Tallman et al.: Заместительная терапия наследственной недостаточности ферментов. Использование очищенной церамидетрихексозидазы при болезни Фабри. В: Медицинский журнал Новой Англии . Том 289, номер 1, июль 1973 г., стр. 9-14, DOI: 10.1056 / NEJM197307052890103 . PMID 4196713 .
- ↑ RJ Desnick, KJ Dean et al.: Ферментная терапия при болезни Фабри: дифференцированный плазменный клиренс in vivo и метаболическая эффективность изоферментов альфа-галактозидазы A в плазме и селезенке. В: PNAS . Том 76, номер 10, октябрь 1979 г., стр. 5326-5330, PMID 228284 . PMC 413135 (полный текст).
- ↑ CA Mapes, RL Anderson et al.: Замещение ферментов при болезни Фабри, врожденная ошибка метаболизма. В кн . : Наука . Volume 169, Number 949, September 1970, pp. 987-989, PMID 4914726 .
- ↑ T. Ohshima, GJ Murray et al.: Мыши с дефицитом альфа-галактозидазы A: модель болезни Фабри. В: PNAS. Volume 94, Number 6, March 1997, pp. 2540-2544, PMID 9122231 . PMC 20124 (свободный полный текст).
- ↑ DF Bishop, DH Calhoun et al.: Альфа-галактозидаза A человека: нуклеотидная последовательность клона кДНК, кодирующего зрелый фермент. В: PNAS. Volume 83, Number 13, July 1986, pp. 4859-4863, PMID 3014515 . PMC 323842 (полный текст).
- ↑ Р. Корнрайх, Р. Дж. Десник, Д. Ф. Бишоп: Нуклеотидная последовательность гена альфа-галактозидазы А человека. В: Исследование нуклеиновых кислот . Том 17, номер 8, апрель 1989 г., стр. 3301-3302, PMID 2542896 . PMC 317741 (полный текст).
- ↑ DF Bishop, R. Kornreich, RJ Desnick: Структурная организация гена альфа-галактозидазы A человека: дополнительные доказательства отсутствия 3 'нетранслируемой области. В: PNAS. Том 85, номер 11, июнь 1988 г., стр. 3903-3907, PMID 2836863 . PMC 280328 (полный текст).
- ↑ Р. Шиффманн, Г. Дж. Мюррей и др.: Инфузия альфа-галактозидазы А снижает накопление глоботриаозилцерамида в тканях у пациентов с болезнью Фабри. В: PNAS. Выпуск 97, номер 1, январь 2000 г., стр. 365-370, PMID 10618424 . PMC 26669 (полный текст).
- ↑ Агалсидаза бета (сухое вещество Fabrazyme®; Genzyme GmbH). Проверено 1 сентября 2011 г.
- ↑ Agalsidase alfa (инфузионный раствор Replagal®; TKT Europe). Проверено 1 сентября 2011 г.